


研究背景
到目前为止,致力于高压LIB的液体电解质工程中的无数尝试主要依赖于调节本体Li+溶剂化结构。通过微调电解质组分,可以在初始循环期间形成稳定的正极-电解质界面(CEI),以减轻高压条件下的连续催化反应。然而,LCO的表面结构不同于体相,并且处理大量的悬空键,这意味着即使在原始状态下,表面结构也固有地不稳定。在充电过程中,表面CoO 2层弯曲和寄生反应的结构变形将更加严重,而没有稳定的CEI层形成来保护高灵敏度表面,从而最终导致EEI退化。因此,设计用于高压LIB的底部溶剂理想地应具有以下特性:i)电解质组分与正极悬挂键之间的强相互作用; ii)固有的抗氧化剂性质以防止严重的电解质分解。然而,合理的溶剂分子选择仍然依赖于典型的描述符,如分子极性指数或静电势,这是基于整个溶剂分子的货车表面性质来评估Li+溶剂化过程。然而,正极表面比静态Li+溶剂鞘层更复杂,导致为溶剂分子的全局性质引入的这些描述符不足以量化EEI上溶剂分子的界面性质。因此,迫切需要解耦溶剂分子与界面电化学性能之间的固有结构-性质关系,以探索适合高压电解质设计的描述符。
成果简介
本文揭示了高压正极界面不稳定性的根本原因与表面过渡金属原子在亚稳四面体间隙中的大量悬空键有关,并提出了一种双描述符定制策略,探索合理的底部溶剂分子与悬空键相互作用,以提高高压条件下固有的正极稳定性。这一策略的关键在于利用底层溶剂分子的固有物理参数来量化复杂的表面吸附和氧化过程,从而探索出一种理想的抗氧化剂底层溶剂分子,该分子能够在致密CEI形成之前与正极悬空键发生强相互作用并微调动态双电层(EDL)。引入Mulliken电荷和Laplacian键级(LBO)双描述符来量化复杂的正极界面性质,并最终选择乙腈(AN)作为构建高压电解质的基底溶剂。AN基电解质与LiDFOB和LiPO 2F 2配合使用,有效抑制了LCO在4.6V贫锂状态下的浮测漏电流,进一步减轻了正极表面的催化作用。同时,用更多的阴离子诱导的离子-溶剂-配位(AI-ISC)结构微调电解质组分实现了阴离子诱导的氟化CEI的形成,以延长LCO寿命。值得注意的是,AN基电解质能够实现0.6 Ah Gr|| LCO软包电池具有超过900次循环的出色循环能力以及80%的容量保持率。利用双描述符理论,我们证明了AN与表面Co─O四面体之间的界面电荷转移对表面Co自旋态的抑制作用,从而有效地抑制了表面能带重叠,减轻了界面晶格氧的不稳定性和晶格应变。这种对界面化学调控的深刻认识将为高压锂电池的发展开辟一条新的途径。该工作以“Dual-Descriptor Tailoring: Rational Solvent Molecule Tuning Enables High-Voltage Li-Ion Batteries”为题发表在Advanced Materials上。
研究亮点
(1) 提出了一种双描述符定制策略,探索合理的底部溶剂分子与悬空键相互作用,以提高高压条件下固有的正极稳定性
(2) 用更多的阴离子诱导的离子-溶剂-配位(AI-ISC)结构微调电解质组分实现了阴离子诱导的氟化CEI的形成
(3) 利用双描述符理论和DFT、MD等理论计算,证明了AN与表面Co─O四面体之间的界面电荷转移对表面Co自旋态的抑制作用,从而有效地抑制了表面能带重叠,减轻了界面晶格氧的不稳定性和晶格应变
图文导读
溶剂评估双描述符的开发
作者首先对涵盖腈、碳酸酯、砜、醚和磷酸盐溶剂的16种典型基底溶剂分子进行了详尽评价。从本质上讲,界面Frumkin效应的吸附溶剂分子源于局部电荷分布,这是由他们的偶极子性质。正如预期,在9个基本物理参数中,选择Mulliken电荷(IMull)作为吸附描述符,并将其与底部溶剂分子和LCO之间的界面相互作用相关联(图1c)。IMull描述符将Janus LCO/电解质界面上复杂的界面相互作用重新表述为吸附溶剂分子的定量参数(Pearson's r:-0.95),从而极大地简化和优化了巨大化学空间中的筛选和计算。
为了实现上述目标,底部溶剂分子必须具有的另一个关键特性是固有的抗氧化性。在大多数研究中,最高占据分子轨道(HOMO)已被广泛用于评估底部溶剂的抗氧化性能,这极大地忽略了其他轨道的贡献,从而导致氧化电位与实验结果显著偏离。考虑到化学键形成的所有轨道贡献,LBO基于在模糊重叠空间中积分电子密度的拉普拉斯算子的负部分,被选择作为另一个描述符,以将分子的物理性质与它们的氧化电位相关联,用于HVE设计(图1d)。正如预期的那样,由于MBO对极性不敏感,LBO与氧化电位的相关性(Pearson's r:0.93)比Mayer键序(MBO,Pearson's r:0.35)更强。使用LBO描述符,也可以量化底部溶剂分子的固有抗氧化性质。因此,利用LBO和Mulliken电荷的双重描述符,可以筛选出具有强吸附性和抗氧化性的理想基底溶剂。
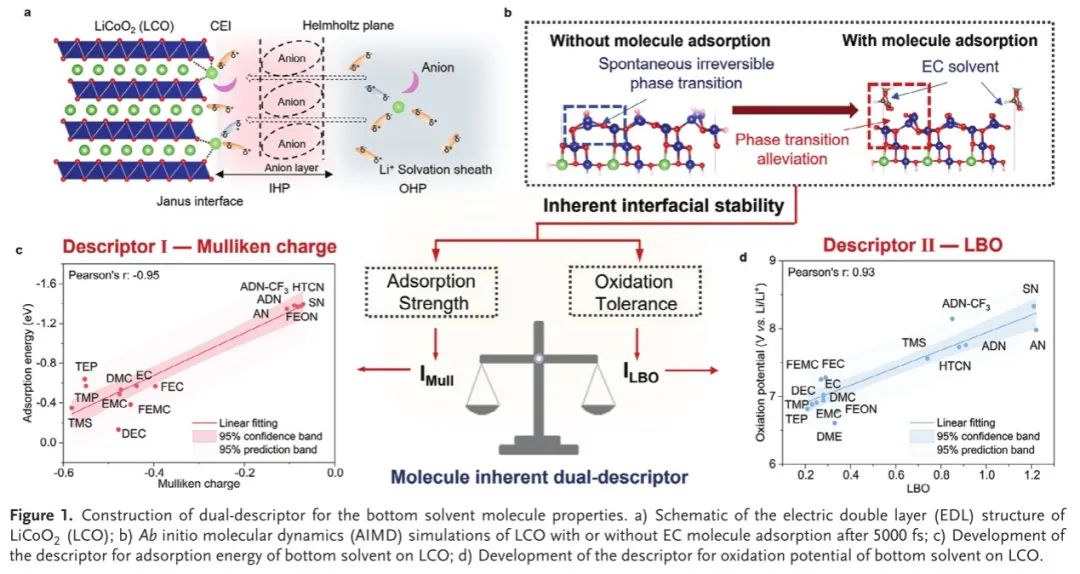
图1.底部溶剂分子性质的双重描述符的构建。a)LiCoO 2(LCO)的双电层(EDL)结构示意图; B)在5000 fs后,对有或没有EC分子吸附的LCO进行了从头算分子动力学(AIMD)模拟; c)开发了LCO上基底溶剂吸附能的描述符; d)开发了LCO上基底溶剂氧化电位的描述符。
在双描述符的指导下,在十六种典型溶剂分子中,最终选择AN作为HVE设计的底部溶剂,这与计算结果一致(图2a)。为了验证双描述符的可行性,我们将AN分子吸附在(104)平面LCO表面上,其中存在比EC更高的电荷转移(图2b),从而减轻放电/充电过程中的解吸。正如预期的那样,在5000 fs AIMD弛豫后,具有底部溶剂AN吸附的Janus LCO/电解质界面保持完整性,表明AN分子在HVE设计中的优越性(图2c)。
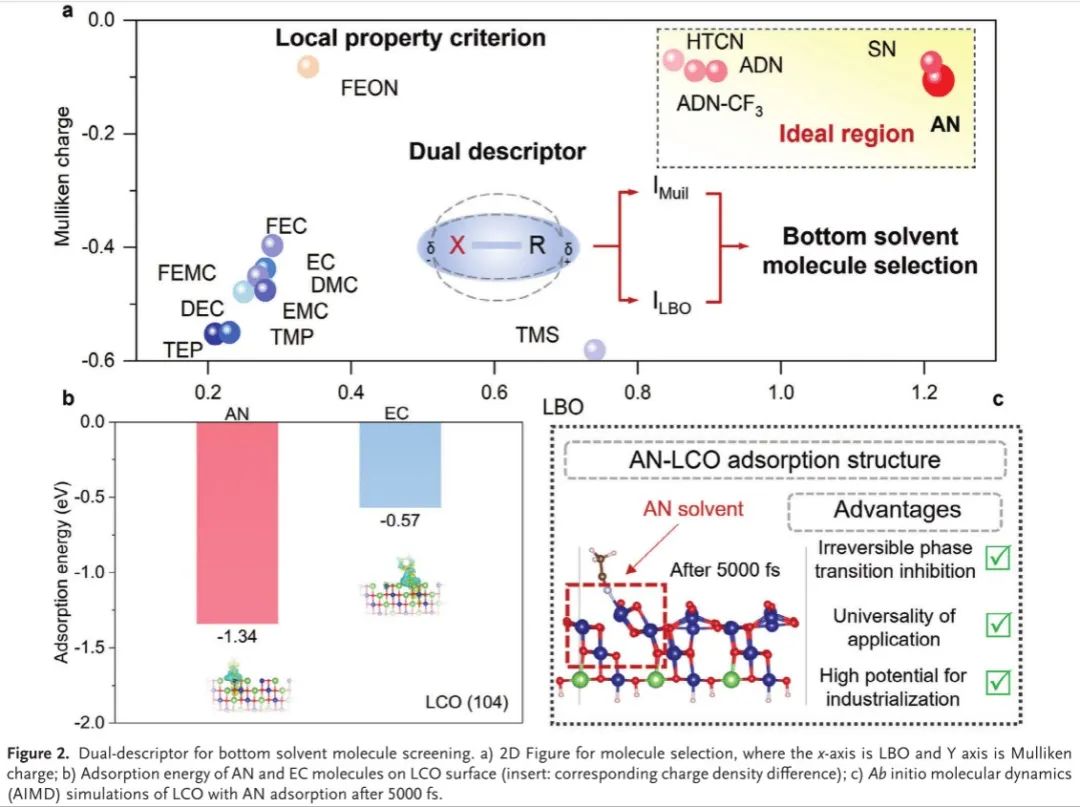
图2.用于基底溶剂分子筛选的双描述符。a)分子选择的2D图,其中x轴为LBO,Y轴为Mulliken电荷; B)AN和EC分子在LCO表面的吸附能(插入:相应的电荷密度差); c)5000 fs后,AN吸附的LCO的从头算分子动力学(AIMD)模拟。
AN基电解质的构建
为了实现HVE的AN底部溶剂的理想溶剂化结构,仔细筛选助溶剂和盐也是必要的(图3a)。近年来的研究表明,加入一定量的低配位数溶剂(LCNS)可以诱导Al-ISC结构的形成,从而形成稳定的富F CEI,并防止电解质严重分解。开始时,作者首先选择LiDFOB作为HVE的主盐,因为它具有较低的最低占据分子轨道(LUMO),并且易于氧化,从而提供富含B/F的无机界面(图3B)。在添加剂方面,还添加了2.5%LiPO2F2以调节SEI和CEI的组分,其在高压下高度稳定。在所选择的共溶剂中,FEC作为典型的LCNS(图3c),最终被选择作为共溶剂以防止在负极侧严重的电解质分解,这也确保了Al-ISC提前氧化分解以诱导富含无机物的CEI层。同时,还选择FEMC作为另一种典型的LCNS作为共溶剂,以稀释高粘度电解质体系(AN/FEC)并诱导更多的AI-ISC结构。最后,作者设计了具有双盐和FEC/FEMC共溶剂的AN基电解质,其可以表示为DEAN(1.0M LiDFOB在AN/FEC/FEMC中,具有2.5%LiPO2F2,Li+/溶剂MR = 1.23:10,AN/FEC/FEMC VR = 1:1:1)。为了进行比较,在这种情况下,选择EC/EMC/DMC中的1.0mLiPF 6作为基础电解质(BE,Li+/溶剂MR = 1:10,EC/EMC/DMC VR = 1:1:1)。
作者研究了Li+在DEAN电解质中的本体溶剂化结构。分子动力学模拟提供了一种简单的方法来表征溶剂化结构和相关的AI-ISC结构的DEAN电解质。显然,尽管在DEAN和BE电解质中盐与溶剂的MR(10)相似,但在DEAN中检测到的AI-ISC结构高于BE。基于先前的研究,我们进行了经典的分子动力学模拟来研究电解质的溶剂化结构。DFOB−和PO 2F 2 −的强径向分布函数峰表明,这两种阴离子都参与了DEAN电解质的内溶剂化框架,平均溶剂化结构为Li(AN)2.5(FEC)1.65(FEMC)0.6(DFOB−)0.75(PO 2F 2 −)0.11。这一结果进一步得到了不同溶剂化构型分布分析的支持,其中大部分阴离子(65%)与DEAN中的Li+配位(图3e)。相反,BE电解质仅提供98%的大溶剂配位比,表明BE中具有较低Al-ISC结构的松散溶剂化结构。通过拉曼光谱进一步验证了DEAN中Li+的溶剂结构(图2 f-h)。当锂盐加入混合溶剂中时,在2278、740和847 cm-1处出现新峰,分别对应于Li+配位的AN、FEC和FEMC峰。此外,阴离子从616 cm−1(游离)蓝移到620和624 cm−1,表明存在CIP和AGG,从而证明了DEAN电解质中以AI-ISC为主的溶剂结构。
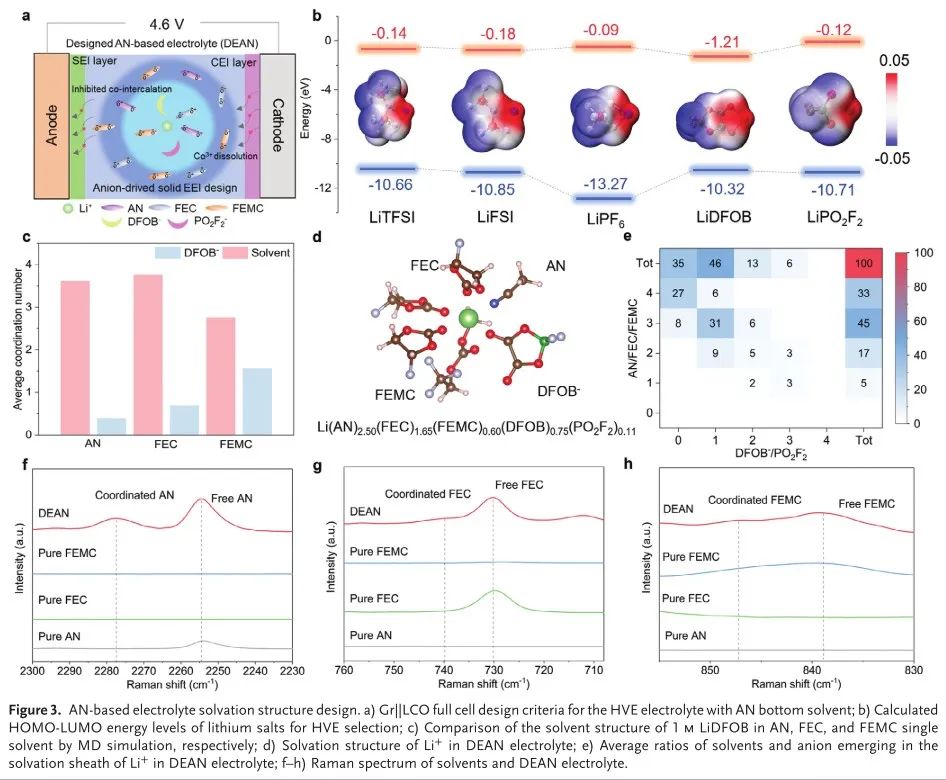
图3.基于AN的电解质溶剂化结构设计。a)级||具有AN底部溶剂的HVE电解质的LCO全电池设计标准; B)用于HVE选择的锂盐的计算的HOMO-LUMO能级; c)通过MD模拟分别比较1 m LiDFOB在AN、FEC和FEMC单一溶剂中的溶剂结构; d)Li+在DEAN电解质中的溶剂化结构; e)DEAN电解质中Li+的溶剂化鞘层中出现的溶剂和阴离子的平均比率; f-h)溶剂和DEAN电解质的拉曼光谱。
4.6V LiCoO2电化学性能的增强
在组装半电池之前,必须评估每种电解质的氧化稳定性。DEAN电解质在4.9 V的高压下表现出显著的耐受性,而BE在4.3 V时表现出显著的漏电流增加。值得注意的是,DEAN在3.5 V时同时提供过氧化过程,归因于具有较高HOMO阴离子(DFOB-和PO 2F 2-)的Al-ISC结构在正极表面分解并形成富含无机物的CEI,与上述计算结果一致。于是,组装每种电解质的LCO半电池以评估LCO在4.6V(相对于Li/Li+)截止电压下的循环稳定性。如(图4a,b)所示,在0.25 C活化3次循环后,每种电解质中的两种LCO在1.0 C下均显示出200 mAh g-1的可逆放电容量(1C = 200 mA g-1)。值得注意的是,在DEAN电解质掺入的情况下,在200次循环后达到90.6%的容量保持率(CR)和99.63%的平均库仑效率(CE)。相反,在BE电解质中,LCO在初始循环中表现出放电电压的显著衰减(图4 b)和比容量的逐渐下降(CR:40.9%,CE:98.07%),表明空白电解质中在高截止电压下发生了严重的不可逆相变。这些发现进一步支持的差异能力的结果。在DEAN中循环的LCO电极的微分电容曲线在200次循环中几乎重叠(图4d)。然而,BE中LCO的dQ/dV峰在200次循环期间逐渐消失,表明显著的结构降解(图4 e)。此外,DEAN电解质与LiNi0.8Co0.1Mn0,1 O2(NCM 811)具有优异兼容性,在4.6 V截止电压下循环200次后容量保持率达80%。除了更好的循环稳定性之外,DEAN电解质还显示出比参比BE更低的过电位增加(图4f),因此证明了对于4.6 V LCO,DEAN相对于商业BE电解质的适用性。为了进一步验证双描述符定制策略的实用性,作者提出了一种Gr|| LCO软包电池(0.6 Ah,N/P比为1.05)。得益于AN底溶剂的强吸附性和抗氧化性,以及DEAN电解液良好的界面钝化能力,4.55 V Gr|| LCO软包电池实现了900次循环的超长工作寿命,具有80.2%的优异CR和99.92%的平均CE(图4g,h)。此外,Gr|| LCO软包电池在900次循环后还提供了80%的显著能量保持率和3.805 V的稳定中值电压,这与之前的报告具有竞争力。
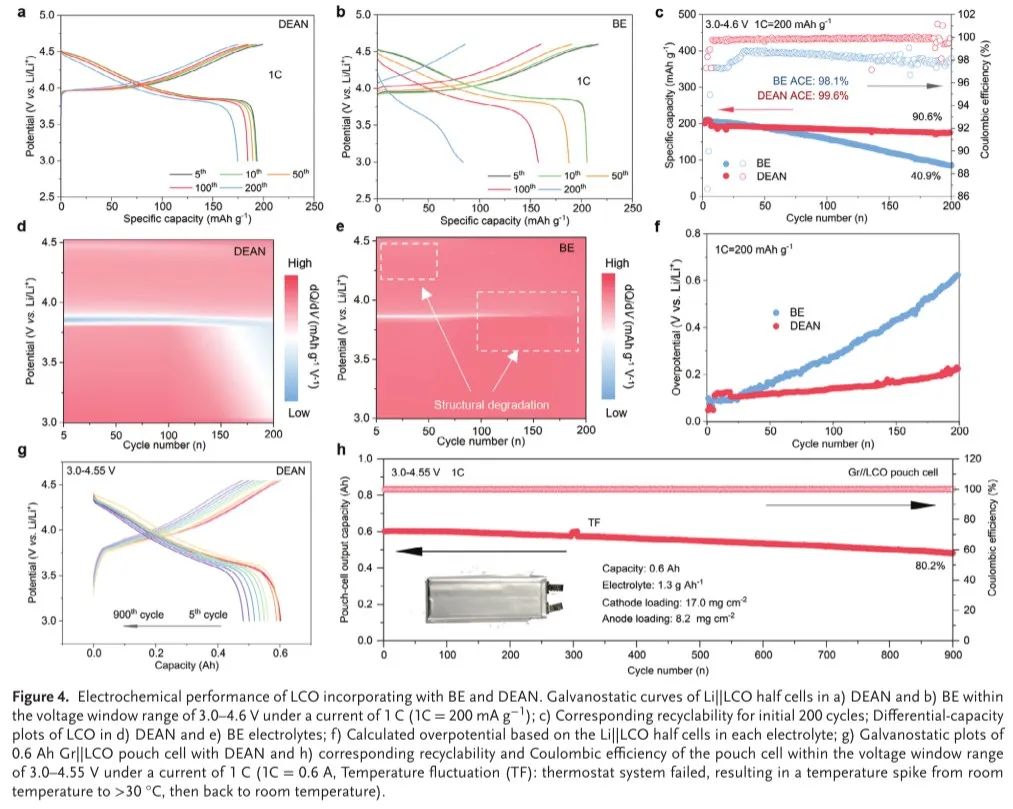
图4.LCO与BE、DEAN复配的电化学性能Li的恒电流曲线||在1 C(1C = 200 mA g−1)的电流下,在3.0 -4.6 V的电压窗口范围内,a)DEAN和B)BE中的LCO半电池; c)初始200次循环的相应可循环性; d)DEAN和e)BE电解质中的LCO的微分容量图; f)基于Li||每种电解液中的LCO半电池; g)0.6 Ah Gr的恒电流图||具有DEAN的LCO软包电池和h)在3.0- 4.55V的电压窗口范围内,在1C的电流下(1C = 0.6A,温度波动(TF):恒温器系统失效,导致从室温到>30 °C的温度尖峰,然后回到室温),软包电池的相应的可再循环性和库仑效率。
结构演化研究
LCO的结构稳定性主要是由从O3到H1-3的初始相变引起的,该相变是不可逆的,具有从表面到体LCO的显著各向异性晶格应变。为了进一步验证这一假设,进行了原位X射线衍射(XRD),以研究在每种电解质的结构演变。(003)的特定峰,其被索引为晶格参数的改变,洞察到充电/放电过程期间LCO电极的相变可逆性(图5a-c)。由于具有Li+嵌入/提取的LCO的c轴的膨胀和收缩,可以在每种电解质中检测到LCO的类似结构演变。但DEAN的(003)峰位移(0.9°)比BE的(003)峰位移(1.8°)小,从而缓解了DEAN中严重的相内应力和结构异质性导致的CoO 2层缓慢滑移。考虑到初始充电过程中CEI层的不完全形成,不可逆结构演化的抑制可归因于底部溶剂AN的作用,其为强吸附,并由于动态EDL结构而能有效抑制应力释放的相变。这一发现得到了485 cm−1(Eg:O─Co─O弯曲)和595 cm−1(A1 g:Co─O伸缩)特征峰的拉曼光谱的进一步支持。在DEAN电解质中(图5d),Eg和A1 g可以可逆地消失/再现并返回到初始强度,而BE中的LCO(图5c)传递了强度的显著降低,这表明在初始循环期间BE中的O ─Co─ O和Co─O键的不可逆断裂和深入的结构崩溃。
在扫描电子显微镜下,使用循环的LCO电极进一步研究了每种电解质中的结构降解。BE电解质中循环的LCO的横截面分析揭示了广泛的开裂(图5 f),而在DEAN电解质中LCO颗粒保持完整(图5e)。这表明,即使在高截止电压下,DEAN电解液也能有效地稳定EEI。高分辨透射电子显微镜(HRTEM)进一步证实了这一发现。我们发现,在DEAN电解液中循环的LCO表面被均匀的CEI层(约5 nm)覆盖,而在循环后,体相LCO几乎不发生岩盐转变(图5e)。相反,BE电解质有一个非常厚的岩盐层(>20 nm),在LCO表面有不均匀的扭曲层涂层(图5 f),表明存在不可逆的O3/H1-3恶性相变。此外,耦合等离子体质谱(ICP-MS)测量也证实了钴的溶解,其显示DEAN中的钴含量比BE电解质中的钴含量降低了30倍(图5g)。这一发现与DEAN中较低的浮动测试漏电流一致(图5 h),表明界面寄生反应随着较低的钴溶解而减少。由于BE电解质中更活泼的表面Co位置,钝化过程应该更快。
然而,快速钝化过程并不意味着电极-电解质界面稳定,快速钝化过程还表明电解质在BE电解液中的强烈分解。此外,BE电解质的强烈分解会在电解质中引入更多的PF 5和HF,从而导致BE电解质中的泄漏电流更高,EEI不断崩溃。另一方面,尽管DEAN电解质中的钝化过程较慢,但阴离子诱导的无机CEI形成可以有效地防止漏电流。更重要的是,这些结果进一步暗示了在DEAN电解质中表面Co位点的活性低于BE电解质,表明在致密CEI形成之前,吸附在LCO表面的AN溶剂的保护。
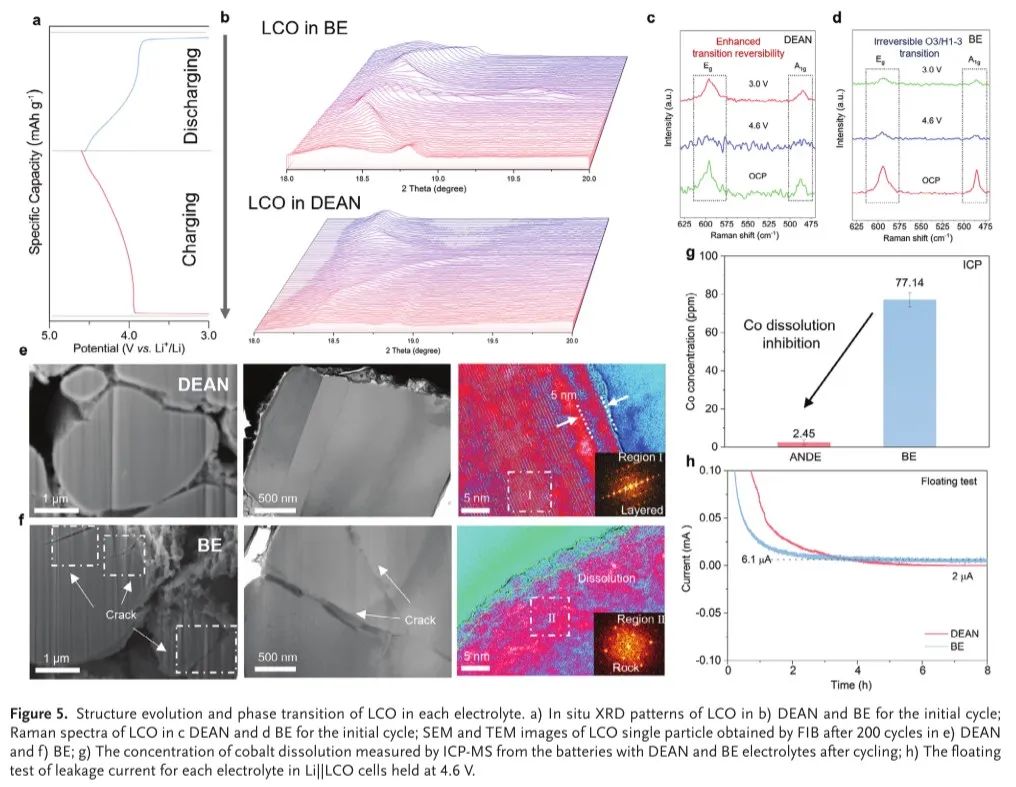
图5.LCO在各电解质中的结构演变和相变。a)初始循环时在B)DEAN和BE中的LCO的原位XRD图案;初始循环时在c)DEAN和d)BE中的LCO的拉曼光谱;在e)DEAN和f)BE中200次循环后通过FIB获得的LCO单个颗粒的SEM和TEM图像; g)循环后通过ICP-MS从具有DEAN和BE电解质的电池中测量的钴溶解浓度; h)每种电解液在Li中的漏电流浮动试验||LCO电池保持在4.6 V。
AIMD计算结果从理论上证明了DEAN的不可逆相变抑制机理得益于AN底部溶剂的选择。然而,基于AN的电解质的机理基础仍然知之甚少。考虑到AN与LCO表面之间的强相互作用,表面上的三配位Co原子可以有效地转变为四配位状态(图6a)。为了理解这种转变,我们转向经典的晶体场理论,该理论假设LCO的键结构强烈影响界面稳定性,直接与表面Co─O配位态有关。在所有能级中,eg * 和t2 g * 是在Co 3 +/4+氧化还原过程中发挥关键作用的关键带,该过程涉及电子去除和填充(图6 b),并且可以用IMull描述符进行量化。Co和O能带之间的重叠对于从O到Co的强烈电荷转移是至关重要的,通常由于晶格应变和失配而导致不稳定的晶格氧。增强界面稳定性需要抑制这种表面带重叠,显着表明,调整高自旋Co状态是必不可少的表面结构。如图6c所示,在费米能级处,表面四面体间隙中的Co原子的态密度为零,表明Co 3+的低自旋构型。在与AN分子相互作用时(图6d),费米能级处的Co原子的态密度表明铁磁性的出现,表明Co向高自旋态的转变。与空白LCO表面结构相比,这种自旋跃迁导致AN吸附的LCO的t2g* 带中的电子密度更高。因此,这减少了充电过程中的Co─O带重叠,有效地抑制了在形成稳定的CEI之前的有害的初始相变。具有高自旋Co原子的LCO可以增强其循环能力,并为高压电解质的发现提供了新的路线图,从而通过双描述符定制筛选用于表面自旋状态调节的溶剂。
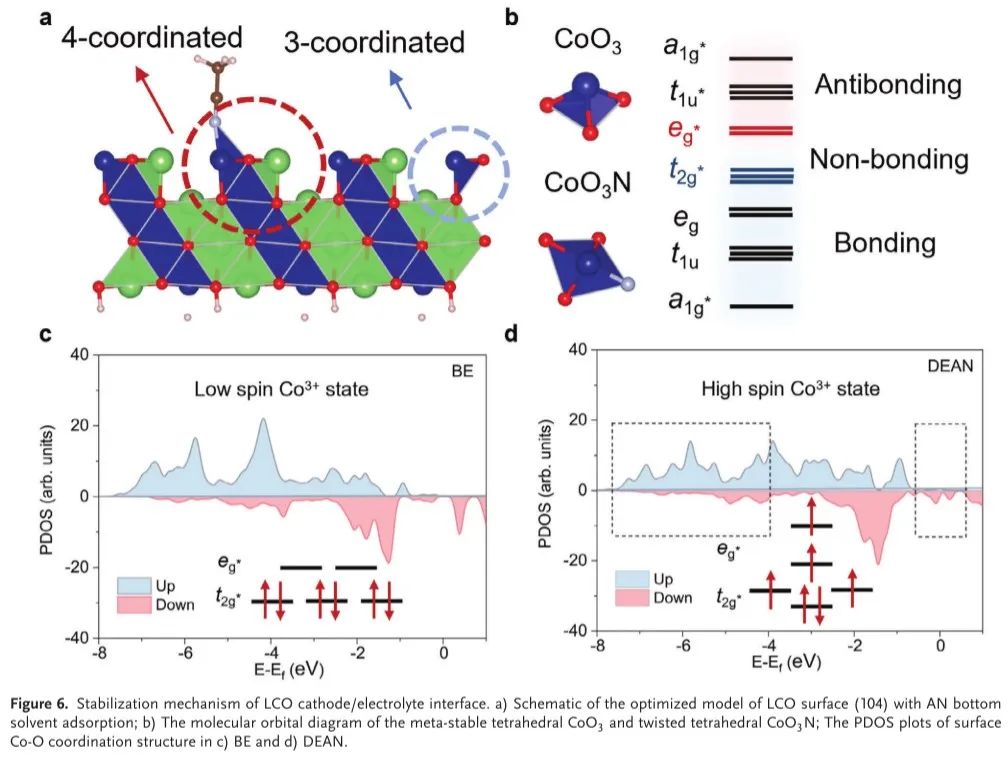
图6.LCO正极/电解质界面的稳定化机理。a)具有AN底部溶剂吸附的LCO表面(104)的优化模型示意图; B)亚稳四面体CoO 3和扭曲四面体CoO 3 N的分子轨道图; c)BE和d)DEAN中表面Co-O配位结构的PDOS图。
诱导富LiF正极/电解质界面
为了进一步证明双参数电解液设计的优越性,用飞行时间二次离子质谱仪(TOF-SIMS)研究了影响LCO电化学性能的另一个关键因素CEI的形成机理和性质(图7a)。与BE系统(图7b)相比,Dean系统(图7c)中LiF2CEI的有效信号得到了增强,特别是在250s的溅射时间内(图7c),这可以被索引到CEI层。这一发现也得到了LiF_2−图谱结果较高的2D表面分布的支持,表明在Dean电解液中形成了富F的−。相反,在Be电解液中,LiF_2−和CoO_2 CEI的分布随着溅射深度的增加而不均匀,揭示了钴的溶解和持久的电解液分解。这一结论与X射线光电子能谱结果一致,后者表明溶剂和阴离子在液碳表面的吸附−脱氟过程(图7d,e)。原子力显微镜(AFM)测量还检查了在Dean和Be中形成的CEI的均匀性和机械稳定性(图7G)。与BE系统中粗糙和破裂的LCO表面相反,Dean中循环的LCO表面更光滑和更均匀,这是由于抑制了钴的溶解和界面重构。此外,在Dean中形成的这种均匀的CEI还获得了更高的杨氏模数(≈28.87 Gpa,图7g)和更低的表面电位(−249.3到−127.9 mV,图7h)。物理性能的显著提高揭示了CEI形成的DeAN的机械稳定性,这得益于正极上致密的EDL结构以及AI-ISC结构的增加,以形成无机的富F CEI结构
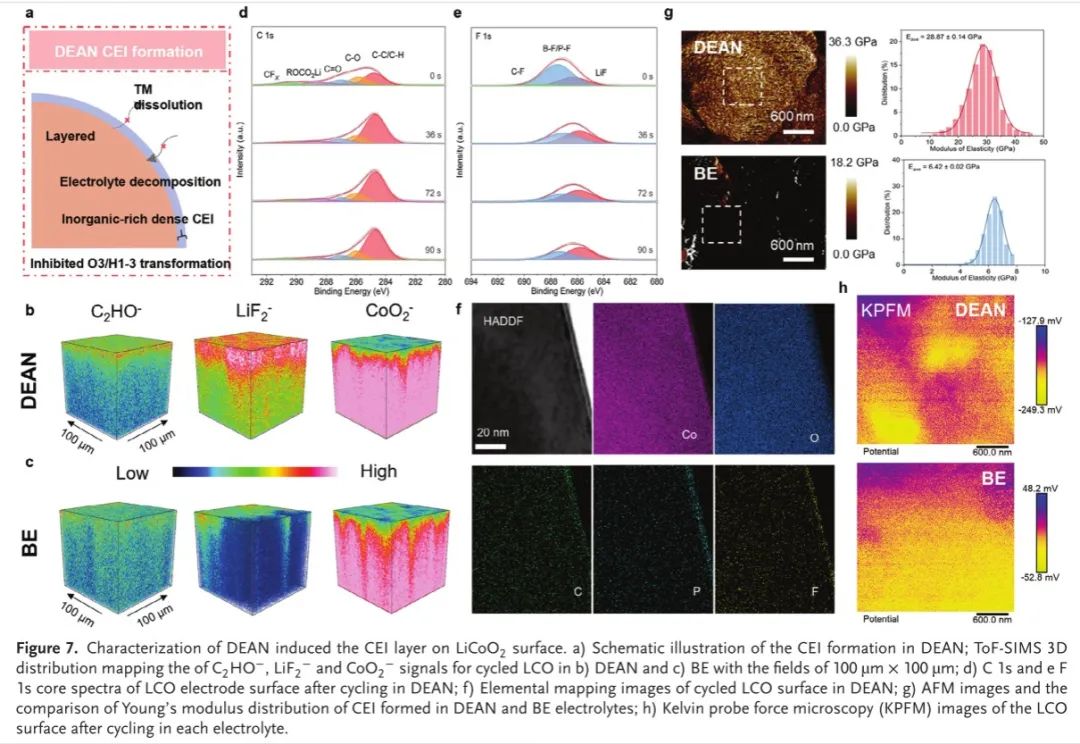
图7.表征了DEAN在LiCoO 2表面诱导的CEI层。a)DEAN中CEI形成的示意图; B)DEAN和c)BE中循环LCO的C2 HO −、LiF 2 −和CoO 2 −信号的ToF-SIMS 3D分布图,场强为100 μm × 100 μm; d)在DEAN中循环后LCO电极表面的C1 s和eF 1 s核心光谱; f)在DEAN中循环LCO表面的元素分布图; g)AFM图像和在DEAN和BE电解质中形成的CEI的杨氏模量分布的比较; h)在每种电解质中循环后LCO表面的开尔文探针力显微镜(KPFM)图像。
DEAN中循环LCO表面上薄且均匀的F元素分布进一步证明了这一假设(图7 f),其本质上抑制了暴露的新鲜表面结构,在高截止电压下弹性模量、硬度和断裂韧性显著下降。通过密度泛函理论(DFT)计算进一步研究了在CEI形成后并入DEAN电解质的超稳定4.6V LCO背后的潜在机制。理论上,当LixCoO 2中的电荷转移到Li0.2CoO2时,电子从Co 3d:t2 g轨道中被提取出来(图5 h),而费米能级随着O 2 p能带顶部以及重叠的Co 3d轨道向下移动。因此,电子将自发地在Co 3d和O 2 p杂化轨道之间转移,导致晶格氧释放以及钴溶解。这种轨道杂化程度可以通过晶体轨道汉密尔顿布居(COHP)来量化,LCO本体表面在费米能级处的−COHP积分值为1.24 eV,表明在贫锂阶段寄生反应的热力学优势。作者接下来研究了在DEAN电解质中形成的CEI,其中溶剂和阴离子结构转化以及LCO表面上的脱附(图8a、B)。如图所示,当来自有机/无机CEI组分的F原子与表面Co原子连接形成Co─O─F八面体时,费米能级处的−COHP积分值显著降低,并削弱了加宽的Co 3d和O 2 p态之间的重叠程度(图8d)。结果,可以显著地抑制不稳定结构和来自晶格氧氧化的外部刺激,从而进一步减轻由氧空位促进的Co迁移和还原。该结果与图6中的晶体场理论分析一致,其进一步证明LCO的界面稳定性由表面亚稳Co原子配位态主导。因此,通过双描述符设计电解质配方,微调表面Co 3+自旋态,提高LCO的固有稳定性,是进一步高压电解质工程的一个有前途的策略。
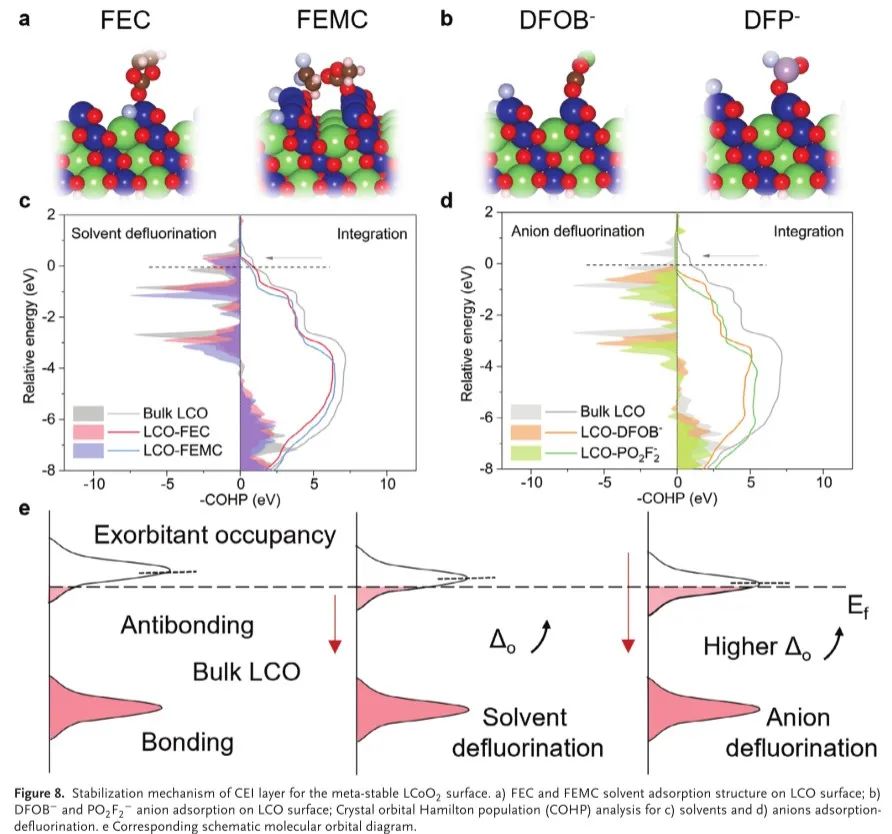
图8.亚稳LCoO 2表面CEI层的稳定机制。a)FEC和FEMC溶剂在LCO表面的吸附结构; B)DFOB−和PO 2F 2 −阴离子在LCO表面的吸附; c)溶剂和d)阴离子吸附-脱氟的晶体轨道汉密尔顿布居(COHP)分析。e相应的分子轨道示意图。
总结与展望
作者提出了一个通用的自上而下的设计概念,高压LCO电解质。采用Mulliken电荷和LBO双指示剂定量筛选吸附性强、抗氧化性强的塔底溶剂。理想的底部溶剂动态地调节EDL功能域,并通过底部溶剂分子和正极悬空键之间的相互作用稳定表面四面体间隙中的亚稳态过渡金属原子。同时,引入合适的助溶剂和成膜Li盐,可以诱导更多的Al-ISC结构以得到致密和薄的富F CEI,抑制表面结构的急剧变化以及伴随Co和O损失的相崩溃和裂纹扩展到本体LCO。AN是一种很有前途的候选物,最终被选为确定的基底溶剂来构建高压电解质。正如预期的那样,AN基电解质有效地抑制了LCO在致密CEI形成之前的初始相变。此外,通过合理的电解液成分调节,在LCO表面形成了一层坚固而薄的富F CEI,进一步抑制了高压下的催化作用,从而赋予了4.6V LCO超稳定性。因此,可以显著地抑制锂钴氧化物在4.6V下的浮置测试漏电流。值得注意的是,AN基电解质能够实现0.6 Ah Gr|| LCO软包电池,循环次数超过900次,容量保持率高达80%。这项工作揭示了界面化学的结构机制与表面钴原子的自旋状态调节。总之,这项工作证明了双描述符在高压电解质设计中的特定作用,这不仅提供了全面的界面结构理解,而且为具有优异稳定性的高压锂电池开辟了一条令人鼓舞的道路。
文献链接:
https://doi.org/10.1002/adma.202417076
锂电联盟会长向各大团队诚心约稿,课题组最新成果、方向总结、推广等皆可投稿,请联系:邮箱libatteryalliance@163.com或微信Ydnxke。
