


Energist 能源学人 2025年04月10日 12:07

第一作者:Sheng Gong
通讯作者:Weihao Gao和Wen Yan
通讯单位:中国字节跳动研究所、美国字节跳动研究所
【成果简介】
尽管机器学习力场 (MLFFs) 在固体和小分子中得到了广泛应用,但在将MLFFs应用于模拟液体电解质方面仍存在显著差距。
对此,来自中国和美国字节跳动研究所的Weihao Gao和Wen Yan等介绍了一个用于分子动力学模拟的预测框架-字节跳动人工智能分子模拟加速器 (BAMBOO),并设计了一个基于物理启发的图形等变变换器 (GET) 架构,作为BAMBOO的核心,以从量子力学模拟中学习,展示了其在锂离子电池液体电解质方面的应用能力。此外,还引入了一种集成知识蒸馏方法,并将其应用于MLFFs,以降低分子动力学模拟中观察结果的波动。最后,提出了一个密度对齐算法,以使BAMBOO与实验测量结果相一致。结果显示,BAMBOO在预测液体电解质的关键性质 (如密度、粘度和离子电导率) 方面达到了最先进的精度水平,这些性质涵盖了各种溶剂和盐的组合。当前的模型已在15种以上的化学物质上进行了训练,在与实验结果相比时,其在各种组成中的平均密度误差仅为0.01 g cm−3。
相关研究成果以“A predictive machine learning force-field framework for liquid electrolyte development”为题发表在Nature Machine Intelligence上。
【研究背景】
液体电解质是锂离子电池的关键组成部分,但其多组分特性使得通过实验优化既昂贵又耗时。原子模拟是替代实验的高效方法,但量子力学模拟计算量大,经典力场精度不足。MLFFs结合了两者优点,展现出快速计算和高精度拟合的特点。MLFFs的发展趋势包括引入图神经网络 (GNN) 等机器学习概念,以及强调基于物理基础的相互作用。然而,液体电解质的MLFFs研究较少,其复杂局部结构 (如溶剂分离离子对、接触离子对和聚集体) 增加了模拟难度。此前的尝试虽有成功,但缺乏普遍适用性证据。此外,MLFFs还面临模拟崩溃、结果波动以及依赖量子力学模拟数据导致的实验结果偏差等问题,限制了其实际应用。
【研究内容】
为了突破这一限制,本研究介绍了一个名为BAMBOO的MLFF框架,它专门用于模拟液体电解质,尤其是在锂离子电池中的应用。文章详细阐述了BAMBOO的设计原理、架构、训练方法以及其在预测液体电解质关键性质方面的性能表现。
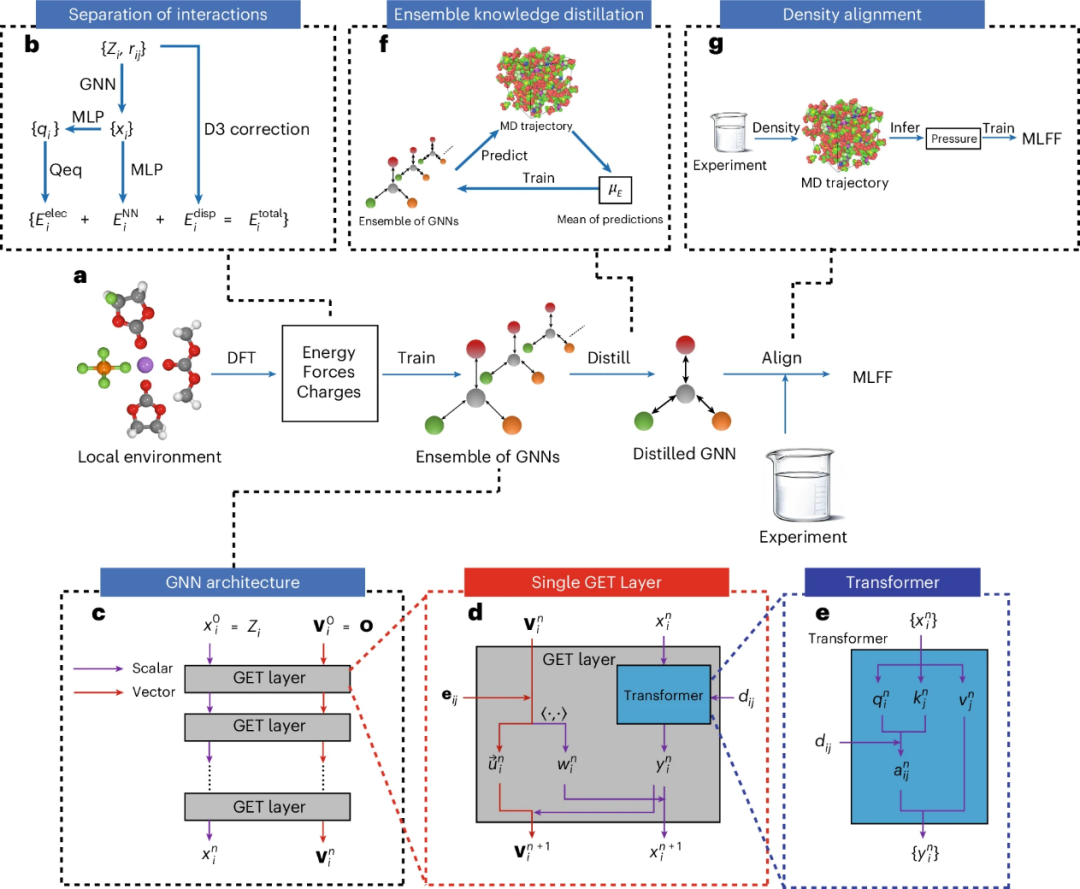
图1. BAMBOO的概述。
BAMBOO的工作流程包括:从液体电解质中采样局部原子环境为气相团簇,通过DFT计算其能量、原子力和电荷。DFT数据集涵盖了多种分子、盐以及锂离子电池中常见的组分,还加入了乙醇、丙酮和Novec 7000等物质,以展示模型的广泛性。利用DFT数据训练多个GNN,再通过集成知识蒸馏融合为一个统一的GNN,最后用实验密度数据对MLFF模型进行对齐。
在能量计算方面,BAMBOO将半局域、静电和色散相互作用分开处理。半局域能量由包含GET层的GNN建模,通过原子类型和相对坐标输入,输出原子表示后预测能量;静电能量通过预测原子部分电荷并计算得到;色散能量基于DFT-D3校正计算。总原子能量由三部分能量相加得到,系统总能量通过对所有原子能量求和计算。此外,基于相对坐标计算能量可自然定义成对力,原子力通过对成对力求和得到,满足牛顿第三定律。
GET架构
GET架构是BAMBOO的骨干,它将原子类型和三维坐标作为输入,并计算能量。GET通过GNN中的变换器层来交换原子间的局部信息,并且能够处理半局部、静电和色散相互作用。这种架构的设计灵感主要来自TorchMD-NET,它通过在图上使用注意力机制,将计算复杂度从O (N2) 降低到O (N),从而提高了效率。
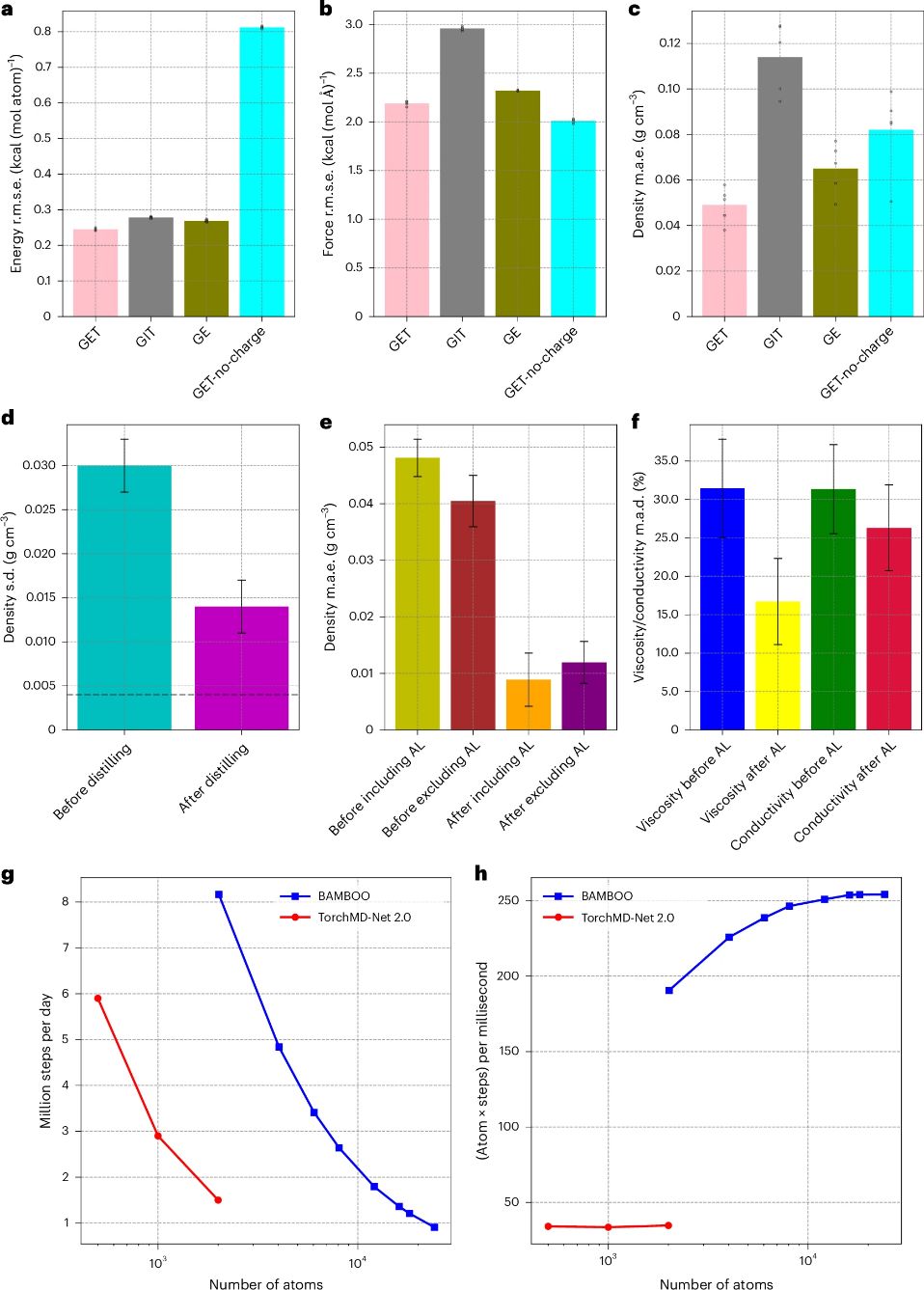
图2. GET层、集成知识精馏和密度对齐的影响。
集成知识蒸馏和密度对齐
由于机器学习中的固有随机性,使用不同随机种子训练的GNN在分子动力学模拟中可能会产生不同的宏观属性,如密度。为了解决这一问题,研究者采用了集成知识蒸馏方法,通过将多个GNN的预测结果取平均值来优化MLFFs,从而显著降低了密度预测的标准差。
为了使BAMBOO的预测与实验数据更好地对齐,研究者提出了一个基于物理的密度对齐方法。该方法利用实验密度来推断MD模拟所需的调整压力,进而调整BAMBOO中的参数。这种对齐方法只需要少量的实验数据,并且具有一定的可转移性,能够改善未包含在对齐过程中的液体的预测。
溶剂化结构和原子部分电荷
BAMBOO能够明确预测原子部分电荷,从而为理解液体电解质中的溶剂化结构提供新的视角。通过模拟不同浓度的LiFSI电解质,研究者分析了Li+和FSI−的电荷分布,并观察到随着盐浓度的增加,溶剂化结构的比例发生变化。例如,在1.12 m、2.25 m和3.74 m的LiFSI浓度下 (m是摩尔浓度,表示每千克溶剂中盐的摩尔数),SSIP (仅由溶剂分子组成的结构)、CIP (包含一个阴离子的结构) 和AGG (由多个阴离子组成的结构) 的比例分别从0.751、0.171、0.078变化到0.182、0.276、0.542。
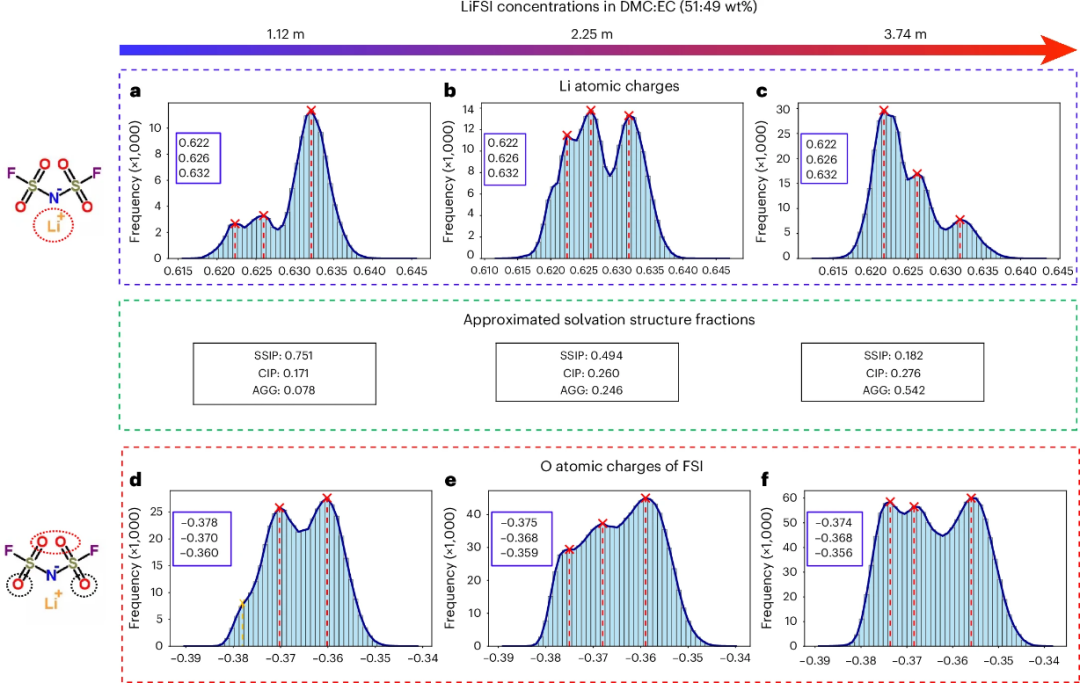
图3. 用BAMBOO模拟三种LiFSI电解质的原子电荷分布和溶剂化结构分数。
BAMBOO框架通过其GET架构、集成知识蒸馏和密度对齐算法,在预测液体电解质的性质方面达到了前所未有的准确性。它不仅能够准确预测密度、粘度和离子电导率,还能够提供关于溶剂化结构和原子部分电荷的详细信息。这些特性使得BAMBOO成为设计和优化实际液体电解质的有力工具,尤其是在需要考虑多种成分的复杂系统中。此外,BAMBOO的高预测能力和可转移性为开发能够模拟大多数有机液体性质和行为的通用MLFF奠定了基础。
【结论展望】
综上所述,尽管BAMBOO在模拟液体电解质方面取得了显著进展,但仍有改进空间。例如,进一步扩展对齐过程以涵盖更多性质,如电导率的直接对齐,可能会进一步提高模型的预测性能。此外,提高MLFFs的可转移性,使其能够稳定地模拟更多种类的分子,也是未来研究的重要方向。
来源:能源学人
【文献信息】
Sheng Gong, Yumin Zhang, Zhenliang Mu, Zhichen Pu, Hongyi Wang, Xu Han, Zhiao Yu, Mengyi Chen, Tianze Zheng, Zhi Wang, Lifei Chen, Zhenze Yang, Xiaojie Wu, Shaochen Shi, Weihao Gao*, Wen Yan* & Liang Xiang, A predictive machine learning force-field framework for liquid electrolyte development,Nature Machine Intelligence, https://doi.org/10.1038/s42256-025-01009-7
锂电联盟会长向各大团队诚心约稿,课题组最新成果、方向总结、推广等皆可投稿,请联系:邮箱libatteryalliance@163.com或微信Ydnxke。

