

点击左上角“锂电联盟会长”,即可关注!


高镍层状氧化物LiNixM1-xO2 (x≥0.9)因其高能量密度和较低的成本而成为汽车电池极具前景的正极材料。然而,在储存过程中,表面碱性化合物的形成和积累阻碍了它们的大规模生产和商业化。本文采用一种经过验证的化学方法,对四种典型正极(LiNiO2 (LNO),LiNi0.95CO0.05O2 (NC)、LiNi0.95Mn0.05O2 (NM)和LiNi0.95Al0.05O2 (NA))在环境空气储存过程中每种残余锂化合物的演变进行了反卷积和量化。此外,通过测量在不同温度下浸出的LiOH浓度来确定水和正极之间反应的活化能。而残余锂和飞行时间二次离子质谱测量结果表明,空气稳定性总体上遵循NM> NA≈NC> LNO的趋势,老化NM表现出最高的电荷转移电阻和最差的电化学性能。原位x射线衍射和扫描透射电镜结果表明,老化NM中存在大面积的尖晶石样M3-xLixO4相,导致颗粒反应非均质性加剧。最后,一步再煅烧法对完全恢复降解正极是有效的。这项工作为克服高镍正极的空气敏感性问题提供了见解。
1介绍
汽车工业的电气化推动了锂离子电池(LIBs)的大规模生产。这就需要电池材料和电极的制造可扩展性和一致性,特别是正极材料,这是目前限制锂离子电池能量密度和成本的因素。高镍(Ni)层状氧化物正极LiNixM1-xO2 (x≥0.9)由于其高能量密度(≥800 Wh/kg)和功率密度而受到越来越多的研究关注。然而,随着Ni含量的增加,高Ni正极的不稳定性问题呈指数级加重,包括高表面反应性、表面残余Li化合物的生长、颗粒粉末化和热稳定性差。虽然人们主要致力于解决循环稳定性差的问题,但高碱度表面残余锂的积累对高镍正极的大规模应用提出了同样严峻的挑战。不幸的是,在理解所涉及的基本原理方面,这方面在文献中得到的关注较少。
目前关于表面残留锂的形成和影响的共识包括几个关键方面。首先,当Ni含量超过70%时,Ni基层状氧化物表现出空气敏感性,并且随着Ni含量的增加,残余Li种类呈指数增长。其次,由于Ni3+的高反应活性及其与周围空气中的水和二氧化碳的反应,产生了包括LiOH和Li2CO3在内的残余Li物质。第三,残锂的形成伴随着氧损失和表面Li/缺氧岩盐相(NiO)的形成,这与残锂本身一起导致了电池阻抗的增长和电化学性能的下降。第四,碱性LiOH和Li2CO3可以对浆液制备过程中使用的聚偏氟乙烯粘合剂进行亲核攻击,导致形成惰性的碳-碳双键和浆液凝胶。
尽管取得了一些进展,但许多基本问题和问题仍未得到解决。例如,在环境储存过程中,不同表面残留锂的演变尚未明确。此外,不同掺杂剂对高镍正极空气稳定性的影响及其机制尚不清楚。此外,正极与水之间的反应动力学以及掺杂剂对反应动力学的影响尚未定量测量。在结构方面,不同成分正极的表面晶格重建途径有待揭示,表面非活性相对正极结构稳定性的影响尚不清楚。最后,能够快速简单地恢复退化正极的策略仍在探索中。
在这项工作中,我们采用了一种验证的滴定方法,系统地监测了四种具有代表性的正极,LiNiO2 (LNO), LiNi0.95CO0.05O2 (NC), LiNi0.95Mn0.05O2 (NM)和LiNi0.95Al0.05O2 (NA)中残余Li化合物的演变。通过精心设计的装置,我们测量了正极和水之间反应的活化能。此外,我们利用高空间分辨率的飞行时间二次离子质谱(TOF-SIMS)和扫描透射电子显微镜(STEM)揭示了剩余锂的层状结构和正极依赖的表面晶格重建途径。同时,基于全面的电池性能数据和原位x射线衍射表征,我们研究了正极依赖的晶格重建途径对电化学性能和颗粒反应非均质性的影响。最后,证明了一种简单的一步再煅烧策略是可行的,可以恢复退化的正极并克服挑战。
2结果与讨论
2.1环境空气贮存过程中正极表面降解机理
为了准确地定量测定不同残留锂物质含量,本研究采用了水基滴定法和甲醇基滴定法,并通过标准添加法进行了可靠性验证(图S1)。基于不同滴定终点的水基和甲醇基滴定曲线的解释见图S2。之后,我们监测了LNO、NC、NM和NA正极在三个月的环境空气储存期间的剩余锂物质积累情况,如图1a和S3所示。值得注意的是,图S3中仅基于水基滴定法获得的总“残余锂”含量包括从正极浸出的LiOH、表面LiOH和Li2CO3。另一方面,总表面残余锂含量是表面LiOH和Li2CO3的总和(图1a)。
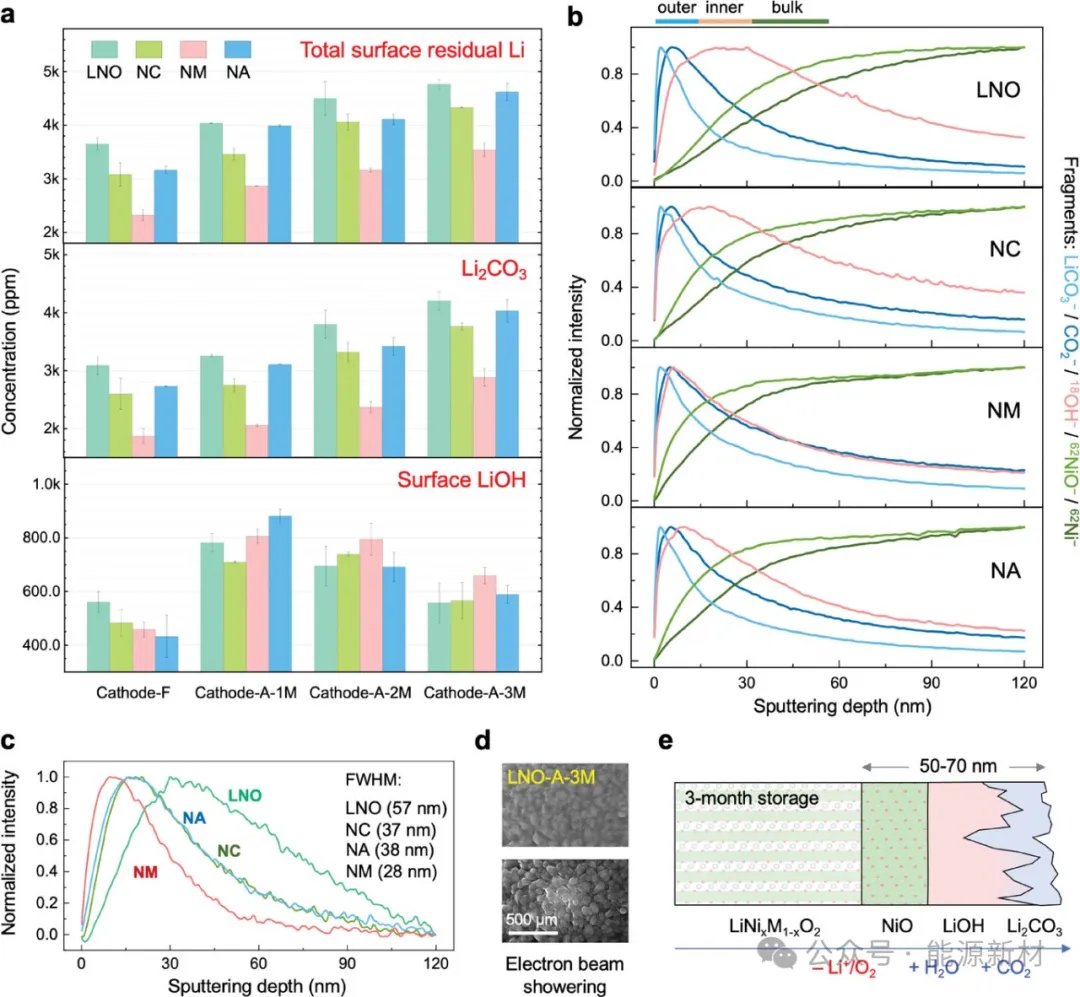
图1a)新鲜正极(正极- F)和老化正极(分别命名为正极-A-1M、正极-A- 2M、正极- A-3M)在不同空气贮存期(每月测量)后表面总残余Li、Li2CO3和表面LiOH含量的演变。b)标准化ToF-SIMS深度分布图和c) LNO、NC、NM和NA正极中62NiO -和62Ni -碎片在空气储存三个月后标准化深度分布图的差异。d)高能电子束激发前后老化LNO正极的TEM图像。e) LNO基正极在空气贮存三个月后表面降解产物的结构示意图。为保证可靠性,在每种条件下对每种样品重复进行两次滴定实验,并给出其平均值。
有趣的是,总“残余锂”含量(图S3)仅在第一个月后增加,然后在所有正极中几乎保持不变。为了理解这种违反直觉的现象,我们进一步研究了浸出的LiOH含量的演变(图S3)。显然,在储存过程中,大块Li+扩散到正极表面并溶解在水溶液中变得越来越困难,导致LiOH浸出受到抑制。这种现象可能是由于表面岩盐相的形成和生长阻碍了Li+的扩散通道,类似于恒流充放电过程中的极化生长。此外,值得注意的是,浸出的LiOH含量与“残锂”含量呈现出不同的趋势。这表明i)不同正极在储存过程中表面岩盐相的厚度是不同的,ii)掺杂剂可能会影响Li+扩散和质子插入的动力学,这将在下面的章节中介绍。
排除浸出的LiOH对剩余锂含量的贡献,基于水和甲醇滴定方法的总表面剩余锂数据如图1a所示。正如预期的那样,每个正极的表面残余锂浓度在存储过程中几乎呈线性增加,并且始终遵循LNO > NC≈NA> NM的顺序。这种趋势与正极中活性Ni3+的浓度密切相关,这被认为是残锂形成的原因。其中,新鲜LNO-F、NC-F、NM-F和NA-F的表面残余Li含量分别为3650、3082、2327和3163 ppm;在这里,字母F是指新鲜的样品。贮藏3个月后,LNO - A-3M、NC- A-3M、NM- A-3M和NA- A-3M的表面残余锂含量分别增加到4764、4330、3542和4621 ppm;这里的A-3M是指在环境空气中储存3个月后。在SEM中可以清晰地观察到正极二次颗粒表面逐渐形成的残余Li,如图S4和S5所示。同时,在储存过程中,氧的损失和Ni3+还原为Ni2+也导致Li+/Ni2+的混合增加(图S6-S9)。
各正极的Li2CO3含量在第一个月呈边际上升趋势,之后呈快速上升趋势,而LiOH含量在第一个月急剧上升,随后逐渐下降。这表明i)新鲜正极与H2O的反应比与CO2的反应更活跃;ii)表面部分LiOH通过与CO2反应转化为Li2CO3; iii)由于LiOH-Li2CO3转化反应,表面残留的Li可能具有层状结构。此外,与表面LiOH相比,表面总剩余锂的变化趋势与Li2CO3更相似。这一观察结果支持了正极与CO2之间的反应可能主要取决于Ni3+含量的假设,而正极与H2O的反应也可能受到其他因素的影响,例如层状结构中Li+和H+的扩散率。
为了进一步了解正极储存后的表面化学性质,采用了具有高空间灵敏度的ToF-SIMS。图1b显示了老化三个月正极的归一化深度分布图。次级离子碎片LiCO3−/CO2−、18OH−、62NiO−和62Ni−分别代表Li2CO3相、LiOH相、富Ni/ O相和含Ni相,其中LiCO3−/CO2−/18OH−和62NiO−/62Ni−分别代表表面残余Li种和本体正极种。深度剖面显示,残锂层呈双层结构,Li2CO3和LiOH分别位于外层和内层。此外,LNO、NC和NA的层状结构比NM的层状结构更突出、更厚。这一观察结果与图1a的滴定结果相吻合,表明Li2CO3的形成部分是由表面LiOH与周围空气中的CO2反应产生的。此外,所有正极上的LiO−(代表所有剩余Li物种)、LiCO3−和18OH−片段的强度比较(图S10)也与滴定结果非常吻合,进一步验证了测量方法。在残锂层下,本体正极也呈现双层结构。具体来说,62NiO−比62Ni−在相互重叠之前更快地达到最大强度。因此,62NiO−可以呈现外表面岩盐NiO相。62NiO−和62Ni−之间的强度差(即I(62NiO−)-I (62Ni−))可以绘制,以评估表面缺锂NiO相的相对厚度,如图1c所示。岩盐层的相对厚度表现为LNO > NC≈NA> NM,这与Ni3+浓度和残余Li含量的变化趋势一致。此外,在更大的尺度上,储存过程中对初级颗粒形态的潜在影响需要澄清。图1d为高能电子束阵雨前后的TEM初级粒子形貌。去除表面残留的锂元素后,初生颗粒形貌保持良好,没有开裂,表明在储存过程中表面晶格氧损失缓慢。
基于以上证据,表面降解机理如图1e所示。在储存初期,由于正极与H2O之间的有利反应,LiOH逐渐在正极的最表面形成。这一过程伴随着表面氧的释放和缺锂NiO层的形成。NiO层的厚度很大程度上取决于正极中Ni3+的浓度,Ni3+的生长抑制了LiOH的形成。在连续贮存过程中,部分表面LiOH逐渐转化为Li2CO3,存在于外层残锂层。在环境空气中储存三个月后,电化学“非活性”层的厚度为≈50-70 nm,其中包括LNO基正极中的NiO, LiOH和Li2CO3。
2.2反应动力学研究
如图1所示,Li2CO3的形成不仅是正极与CO2反应的结果,也是LiOH-CO2反应的结果。此外,与反应活性高度依赖于Ni3+浓度的正极- CO2反应不同,LiOH的生成既来自于正极- H2O氧化还原反应,也来自于Li+-H+交换反应,这使得正极- H2O反应活性可能取决于Ni3+浓度以外的因素。因此,对正极与H2O和CO2的反应进行解卷积并研究反应动力学是至关重要的。
如图2a所示,我们设计了一个实验装置,允许正极-水反应在不同的温度下发生。图2b显示了两种可能的反应机理,其中(1)和(2)分别为氧化还原反应和Li+-H+交换反应。尽管关于哪种反应途径是主导机制的争论仍然存在,但正极和水之间的反应本质上都是刘易斯酸碱反应,正极和水分别是碱和酸。为了量化生成的LiOH ([LiOH]浸出液)的浓度,我们定义了一个想象的LiOH浓度,命名为[LiOH]IM。[LiOH]IM是从正极表面溶解的LiOH ([LiOH] surface)、表面Li2CO3 ([Li2CO3] surface)的浓度和水溶液中的[LiOH]浸出的总和,其中[LiOH] surface和[Li2CO3] surface是一个正极的常数。[LiOH]IM可以很容易地根据水基滴定的第二个滴定终点计算出来(图S2),它只是[LiOH]Leach的函数。
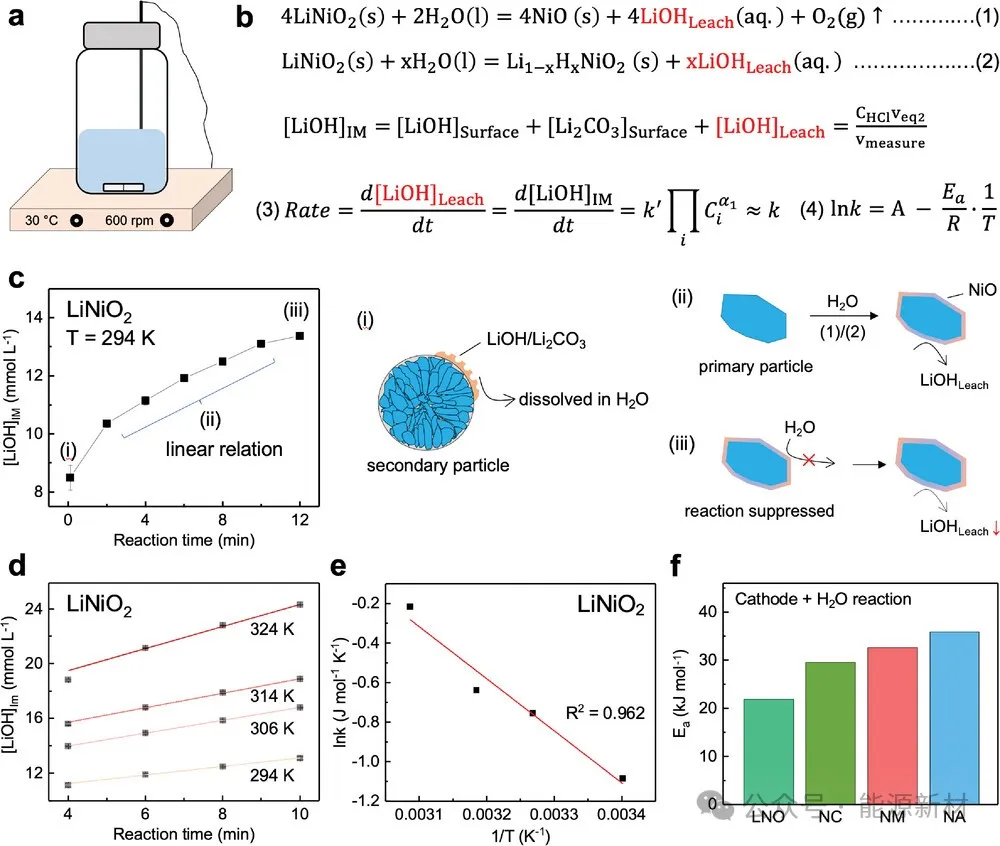
图2a)控制正极与水反应温度的实验装置示意图。b)正极与水之间的反应,以及假想LiOH浓度([LiOH]IM)、反应速率常数和活化能的计算公式。c) [LiOH] IM -反应时间图及正极颗粒与水反应示意图。d)不同温度下LNO正极的[LiOH] IM反应时间图。e) LNO与水反应的Ink -1/T图。F)四个正极与水反应的活化能。为保证可靠性,在每种条件下对每种样品重复进行两次滴定实验,并给出其平均值。
图2c绘制了[LiOH]IM随反应时间的函数图。可以看出,[LiOH]IM曲线呈现出三步特征:1)开始时呈非线性增长,2 ~ 10 min呈线性增长,12 min后呈非线性增长。阶段1)主要表现为表面残余Li溶解过程,阶段2)为正极颗粒与H2O的反应。由于表面岩盐相的形成和溶液中LiOH浓度的增加,抑制了与H2O的正极反应,导致12 min后[LiOH]IM呈非线性增加。
[LiOH]IM与时间的线性关系表明LNO与H2O的反应是零级或准零级反应。的确,如图2b所示,由于固体反应物和生成物的浓度可设为一,水的浓度因其过多而可视为常数,且反应高度不可逆,因此反应的速率定律可以简化为零级模型(速率= k)。根据这一理论,我们测量了不同温度下线性反应区的斜率(即反应常数k),如图2d所示。值得注意的是,在294 ~ 324 K的所有温度范围内,[LiOH]IM和反应时间表现出可靠的线性关系(图S11)。根据图2b式(4)所示的ArrheNius方程,拟合Ink - 1/T曲线即可得到活化能Ea,其斜率为- Ea/R(图2e)。注意,测量到的Ea是图2b中(1)和(2)两个反应的有效反应势垒。使用这种方法,我们测量并绘制了图2f中四个正极的Ea。如图所示,LNO、NC、NM和NA的Ea值分别为22、29、32和36 kJ/mol,与图1a中新鲜正极表面LiOH的浓度非常吻合。注意,这种趋势不能扩展到老化正极,因为表面晶格劣化会抑制与H2O的反应。此外,尽管NC或NM的Ni3+浓度与NA相同或更低,但NA表现出更高的Ea,这表明Al掺杂可以抑制Li+-H+交换反应。
2.3正极再煅烧再活化
为了恢复老化的正极材料,我们采用了一种一步重烧的方法来重新激活老化的正极,而不需要额外的锂源,如图3a所示(也见实验部分)。从本质上讲,表面缺锂岩盐层可以在O2气氛下与剩余的锂物质发生反应,这已经被证明是热力学上可以实现的。注2讨论了支持这种再煅烧机制的进一步实验证据。
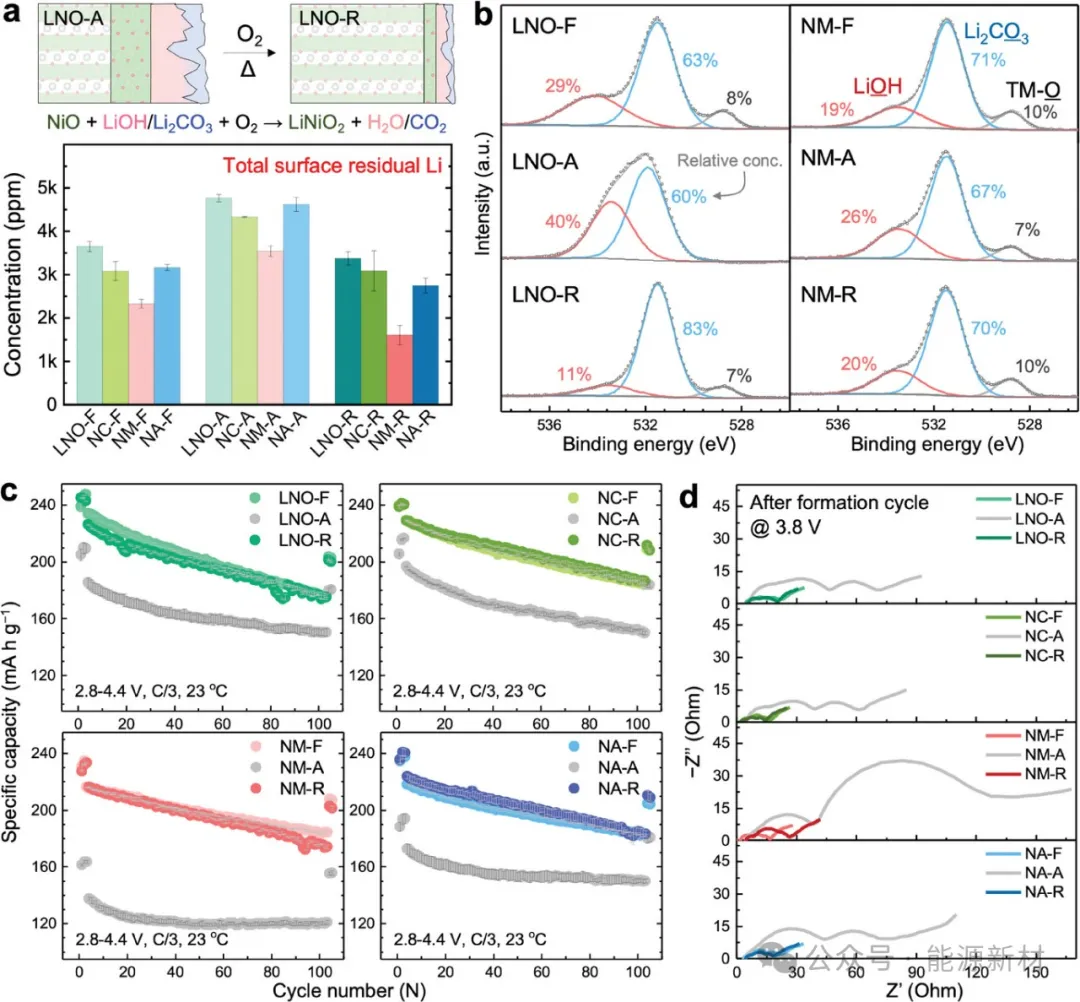
图3a)老化正极再煅烧处理示意图及新鲜、老化(空气贮存3个月)和再煅烧(正极- R) LNO、NC、NM和NA正极表面总残余锂含量。b)新鲜、老化和再煅烧LNO和NM正极的XPS O - 1s光谱。c)新鲜、老化和再煅烧LNO、NC、NM和NA的循环性能和d) EIS图。细胞以2.8 ~ 4.4 V\的C/3倍率循环,C = 180 mA/g。电化学性能数据是基于三个或四个并联电池的平均值。
用残余锂滴定法和XPS测量来评估表面残余锂是否成功地重新插入正极。如图3a所示,与老化正极相比,再煅烧正极的剩余锂含量明显降低。有趣的是,它们甚至低于新鲜正极,可能是由于在再煅烧过程中Li蒸发损失。为了进一步验证表面含锂物质的再插层,我们收集了两种具有代表性的正极LNO和NM的XPS结果,如图3b和图S12所示。正如预期的那样,LNO和NMS光谱与新鲜样品相似,表明表面锂离子的重新嵌入和正极表面化学的恢复。老化正极和再煅烧正极的XRD和SEM图像进一步验证了正极的结构和形态恢复(图S4-S9)。
为了揭示老化和再煅烧对电化学性能的影响,首先在同一批正极的基础上收集新鲜、老化和再煅烧正极的循环性能(图3c)。每个样品至少测试三个平行细胞以确保可重复性。如图3c和图S13-S17所示,所有老化正极都表现出较大的容量衰减较低的第一循环库仑效率。令人惊讶的是,虽然NM表现出最少的剩余锂积累,但从NM- f到NM- a的容量下降比其他正极明显得多。具体来说,NM-A只能提供165 mAh/g的容量,相当于NM-F (234 mAh/g)容量的71%;而LNO-A、NC-A和NA-A的容量分别为211、217和196 mAh/g,与它们各自的新鲜正极相比,其容量保留率分别为87%、90%和84%。NM-A的最大容量损失归因于存储后显著的电荷转移电阻增长,如图3d中的EIS图所示。由于同样的原因,NM-A也比其他老化正极表现出最差的倍率性能(图S18)。
此外,再煅烧正极的循环性能与新鲜正极几乎相同,特别是NC和NA。尽管最初的容量损失很小,但LNO-R似乎比LNO-F具有稍好的循环稳定性。相比之下,NM-R是唯一表现出比NM-F稍差的性能和明显更大的电荷转移电阻的正极。总体而言,电化学性能数据表明,尽管NM具有最佳的空气稳定性,但在环境空气储存和再煅烧后,其阻抗增长在所有正极中都是最高的。
2.4体相和表面结构稳定性演化
NM-A的反直觉电化学行为表明,表面残余锂含量并不是导致其电化学性能下降的主要因素。研究认为,纳米粒在储存过程中不同的结构重构途径可能是其主要原因。为此,首先在现场收集了NM-F、NM-A、LNO-F和LNO-A正极的XRD数据,通过监测充电过程中的体相转变,比较了正极的结构稳定性和反应非均质性。
如图4a,b所示,尽管新鲜和老化正极都经历了H1-H2-H3相变,但老化正极的H2-H3相变发生在较低的荷电状态(SOCs,即衰减程度)下。此外,与NM-F相比,NM-A在高SOC区域表现出H2-H3相分离行为,如图4b中的箭头所示。这表明纳米- a反应具有明显的非均质性。此外,基于(003)和(110)峰位置,可以计算出c和a晶格参数,以量化单元胞中的体积变化(图4c;图S19)。可以看出,NM-A和NM-F的H2-H3相变分别从约70%和76%的SOC开始。在充满电状态下,NM-A的c晶格参数为13.55 Å,明显小于NM-F的13.8 Å。更重要的是,即使在充电结束时,NM-A的H2相仍然存在(图S20);而NM-F的H2-H3相变在80% SOC时完成。在LNO正极上,老化样品比新鲜样品有更大的相分离和更大的晶格参数变化,但程度要温和得多。这与电化学数据非常一致,其中NM在存储后比LNO遭受更多的容量损失。
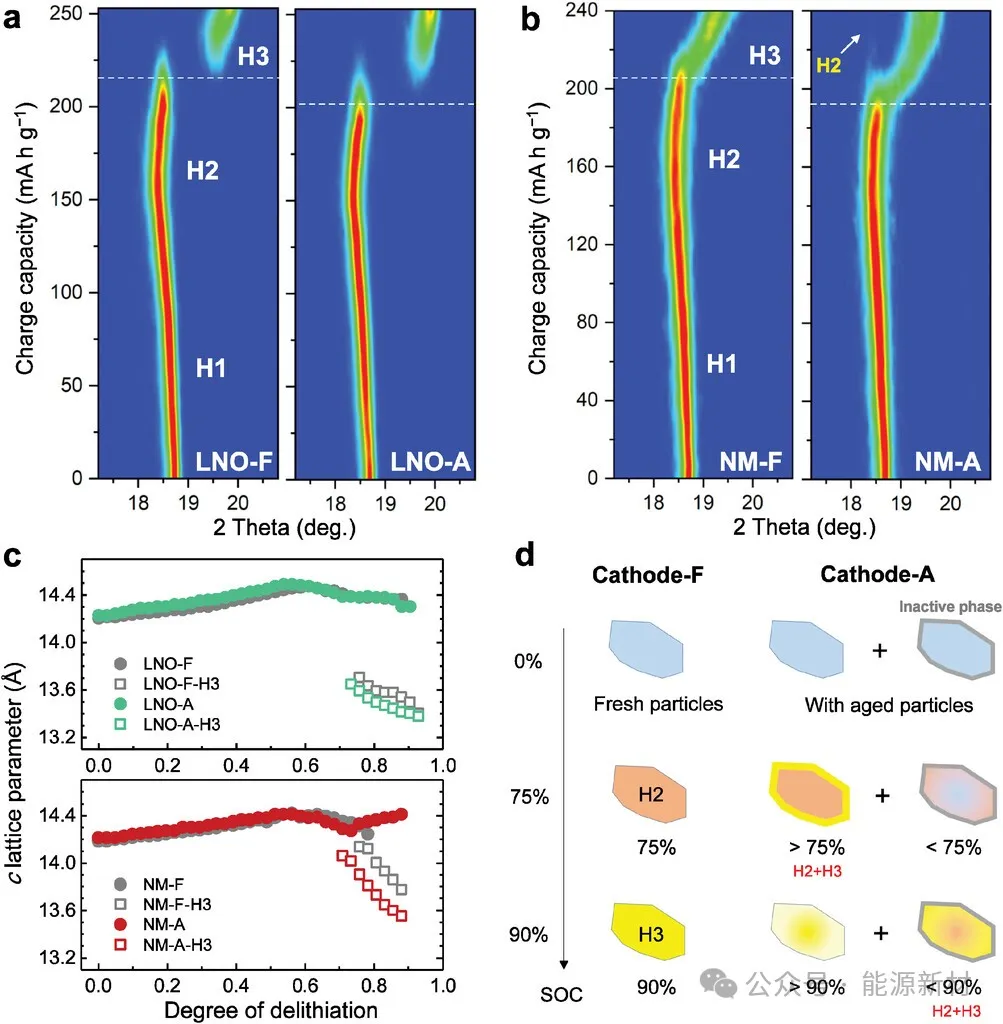
图4a)新鲜和老化的LNO和b)新鲜和老化的NM正极的(003)衍射峰的原位XRD等高线图。c)新鲜和老化LNO和NM正极在充电过程中c晶格参数的演变。d)充电到不同SOC时,新鲜和老化正极中正极初级粒子的衰减状态示意图。
为了解释老化正极H2-H3相变发生早、结束晚的原因,图4d给出了相变示意图。具体来说,老化正极含有表面(或整体)缺氧非活性相(标记为灰色外壳)的初级颗粒,这可能会阻碍Li+在衰减过程中的扩散。因此,当正极充电至75%荷电状态,即将发生H2-H3相变时,老化正极中降解颗粒处于< 75%的SOC,而剩余颗粒处于>75%的较高SOC,开始经历H2-H3相变。这解释了老化正极的H2-H3相变较早发生的原因。当正极充电至90%荷电状态时,新鲜正极的H2 - h3相转变完成,老化正极中降解颗粒的荷电状态< 90%,剩余H2相;另一方面,剩余颗粒的SOCs >90%。这解释了老化正极在充电结束时保留H2相和较大的晶格畸变。总体而言,原位XRD结果表明,虽然Mn掺杂能够增强正极结构的稳定性,但在长期空气储存后,会导致反应动力学更缓慢,反应非均质性更大。
NM-A相对于LNO-A的反应非均质性更大可能是由于掺杂剂影响了不同的晶格重建途径。因此,我们收集了高分辨率的STEM数据来研究LNO-A和NM-A的相形成。如图5a所示,LNO-A被一层厚约6 nm的岩盐NiO相覆盖。有趣的是,在图5b的NM-A中可以识别出大面积的尖晶石样相,而不是岩盐相。再煅烧后,LNO- R表面NiO相可以明显缩小到只有1-2 nm(图5c),与合成后的LNO相相当;相比之下,在NM-R正极中仍然存在小面积的尖晶石样相(图5d),这可能是由于尖晶石样相的结构稳定性较高。这就解释了图3c中NM-R的电化学性能略差于NM-F的原因。
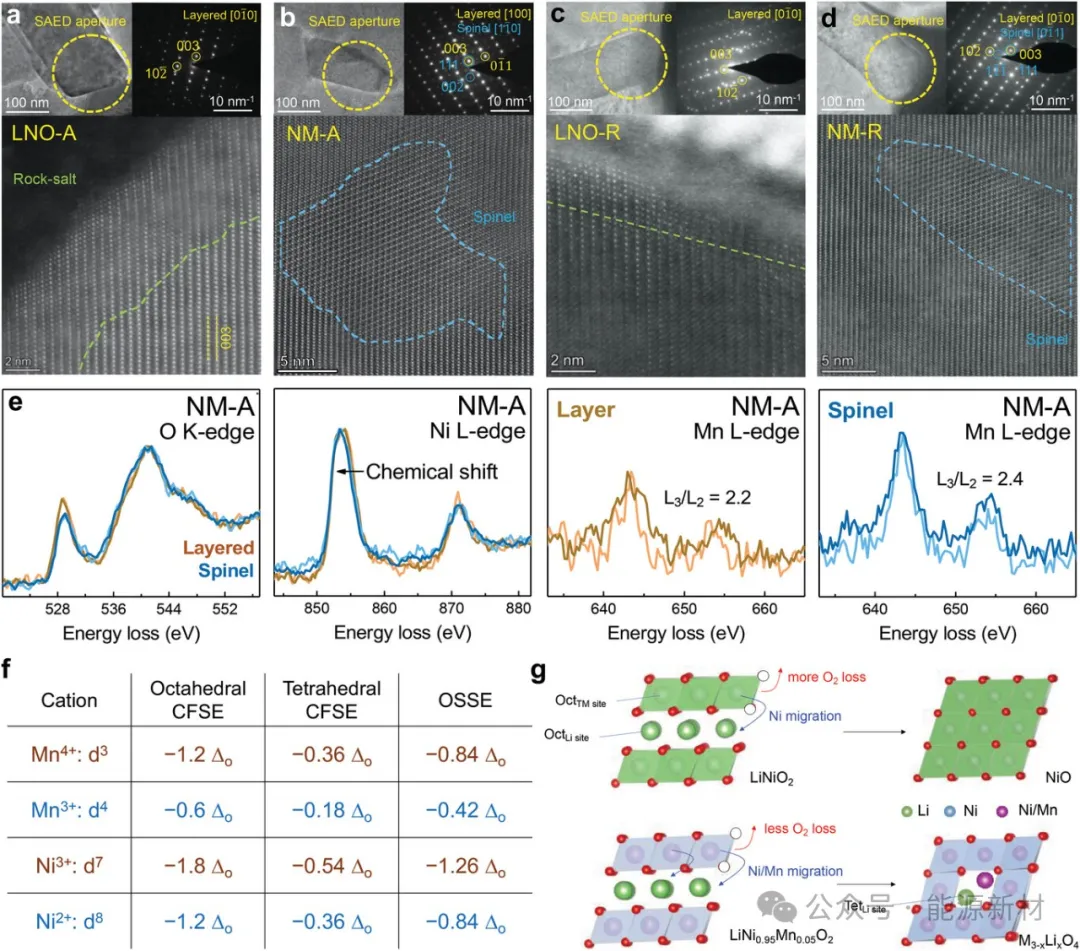
图5a) LNO-A, b) NM-A, c) LNO-R和d) NM-R的选定区域电子衍射和STEM图像。e) NM-A正极层状相(橙色)和尖晶石样相(蓝色)的O k边、Ni l边和Mn l边EELS图。每个地区测试了两个地点。f) Mn4+、Mn3+、Ni3+和Ni2+阳离子的八面体晶体场稳定能、四面体晶体场稳定能和八面体位点稳定能。g) LNO和Mn掺杂NM正极的表面晶格重构路径示意图。
进一步了解类尖晶石相的组成及其形成机理。利用STEM-EELS比较了TM在NM-A的层状相和尖晶石相中的氧化态,如图5e所示。首先,层状相和尖晶石相的O - k边缘的前峰是由于O - 2p和TM - 3d轨道在六配位环境中的杂化所致。与层状相相比,尖晶石相的前峰(529 eV)与O k边峰(542 eV)的强度比更小,表明氧损失导致邻近的TM离子减少。其次,尖晶石相的Ni L3峰位于比层状相更低的能量损失位置,这证实了O k边数据,进一步证明了TM价态的减少。对于Mn l边数据,由于Mn浓度低,Mn l边峰非常弱。这对比较不同位置和样本之间的峰位置提出了挑战。因此,用L3和L2峰的面积比来判断Mn的价态。尖晶石区域的L3/L2比略大表明形成了具有较低Mn价态的类尖晶石相。更重要的是,尖晶石区Mn的l边峰要比层状相强得多,这与表S1中的EDS(能量色散x射线光谱)结果相一致,证明了类尖晶石相中Mn的浓度较高,这是由于Mn的迁移和富集。根据晶体场理论得到的八面体位点稳定能(OSSE)值表明,Mn比Ni更容易从原来的八面体位点迁移到四面体位点(图5f)。
总体而言,电化学性能数据、STEM-EELS光谱和OSSE计算表明:1)TM的价态总体降低;ii)与Mn相比,Ni的价态可能更还原;iii)类尖晶石相中Mn含量高于层状相;iv)尖晶石相具有高电阻性,可能是由于TM离子部分占据了四面体Li位点。根据以上证据可以推测,类尖晶石相的结构介于Li(Mn,Ni)2O4和(Mn,Ni)3O4之间,可以写成M3-xLixO4 (M = Mn和Ni)。大多数可能的成分包括Ni和Mn的非积分含量,如Mn0.15Ni2.35Li0.5O4。由于NM-A中某些尖晶石相的形成不需要像LNO-A中NiO还原那么多的Ni,因此即使在储存过程中氧气和Li的损失较少,NM-A中也会有更多的尖晶石相形成(如图5g所示)。这显著增加了电荷转移阻抗、反应不均匀性和容量损失,如图3、4所示。
3结论
在这项工作中,我们采用了一种经过验证的水/甲醇滴定法来监测LNO、NC、NM和NA四种具有代表性的正极上剩余锂的生长,以了解不同常见掺杂剂在高镍正极中的作用。结果表明,表面残余Li含量、Li2CO3含量和Ni3+浓度均遵循LNO > NC≈NA> NM的规律。然而,由于氧化还原反应和Li+-H+交换反应都可能发生,LiOH含量的变化趋势不同。通过分析LiOH浓度增长随反应时间和温度的变化规律,可以量化正极与水反应的活化能,反应活化能的变化趋势为LNO < NC< NM< NA。这种趋势与Ni3+浓度趋势的偏差是由于Li+-H+交换反应动力学的差异。基于先进的表征,我们证明了剩余的Li物种呈现层状结构,LiOH和Li2CO3分别位于内层和外层,这是由于LiOH转化为Li2CO3。虽然纳米正极在储存后的剩余锂积累最少,但与新鲜正极相比,它的阻抗增长、容量损失和反应非均质性最大。高分辨率STEM数据显示,LNO中的氧损失会在表面形成厚度为6 nm的NiO相,而由于过渡金属离子的减少和Mn从过渡金属层向Li层的迁移,在大部分NM中形成大面积的M3–xLixO4样尖晶石相。最后,我们证明了一步重烧策略在完全恢复正极结构和性能方面的有效性,特别是对于CO和Al掺杂的NC和NA。
在高镍正极的未来发展中,细致的成分设计包括精确的掺杂选择将对不同的应用场景至关重要。基于本研究,提出尽管Mn可以减缓残余Li的形成,但高Ni、高Mn含量的正极需要更合理的储存,以避免形成抗性尖晶石样相。引入小剂量的CO和Al可能有助于减缓尖晶石相的形成,尽管它们在抑制残余Li形成方面不如Mn有效。未来的研究应进一步探讨B等其他有前景的掺杂剂在调节正极空气稳定性中的作用以及掺杂剂相互作用的影响。更重要的是,进一步的研究应该集中在描述氧化还原反应和Li+-H+交换反应对LiOH形成的贡献。此外,活化能测量表明,Al掺杂NA对质子攻击具有最高的抵抗能力。因此,当使用具有活性质子的液体介质中的表面处理作为正极时,Al掺杂可能是首选的选择。最后,本研究中开发的方法,如表面碱性物质的定量,活化测量和再煅烧处理,可以扩展到各种其他氧化物正极材料,包括钠和钾过渡金属氧化物正极。
