



几十年来,锂离子电池(LIB)的进步正在推动全球向电动汽车和电网储能过渡,以补充间歇性可再生能源发电。与此同时,不断升级的锂电池已经使关键电池材料价格紧张,导致电池价格今年首次上涨,扭转了长达十年的价格下降趋势。为了满足在不久的将来不断增长的太瓦时规模的需求,锂离子电池将需要提高其性能,同时变得更便宜,特别是商业锂离子电池正极(例如,层状氧化物LiNi0.6Mn0.2Co0.2O2)中需要对昂贵的Ni和Co寻找替代品(例如,层状氧化物LiNi0.6Mn0.2Co0.2O2),正极成本占目前锂离子电池总材料成本的近50%。
通过提供比层状氧化物(~770 Wh/kg-AM)更高的能量密度(>900 Wh/kg-AM,活性材料AM),同时具有LiFePO4(~580 Wh/kg-AM)的低成本,Mn基阳离子无序岩盐(Mn-DRXs,例如Li2MnO2F, Li2Mn1/2Ti1/2O2F和Li1.68Mn1.60O3.7F0.3)由比镍和钴更便宜的金属(锰、钛等)制成(图1a),是为数不多的无Co/Ni正极材料之一,具有最低的储能成本($/Wh),可以改变目前的正极市场份额。这些材料的良好性能只在含有大量炭黑(15-30 wt%,CB)和粘合剂(5-10 wt%,如聚偏二氟乙烯,PVDF)的高度稀释正极膜中得到证明。在稀释的薄膜中测试AM,会掩盖在CB/粘合剂含量最低的实际薄膜和电池中明显存在的问题(例如,~2 wt% CB, ~2 wt%粘合剂),即使AM水平的问题得到解决,这些问题也会持续存在并限制材料在实际LIB中的使用。此外,稀释电极限制了电极或电池水平的实际能量密度增加(Wh/kg电极/电池,Wh/l电极/电池)(图1b)。例如,Li1.68Mn1.60O3.7F0.3可以提供近1100 Wh/kg-AM,但仅在70 wt%-AM薄膜中(AM仅占电极的~70 wt%),导致正极能量密度为~770 Wh/kg-正极,比商用Ni/Co基层状正极(~740 Wh/kg-正极,~770 Wh/kg-AM)在少量CB和粘合剂的情况下稍高。

近日,蔚山国立科学技术研究院Dong-Hwa Seo教授和麦吉尔大学Jinhyuk Lee教授,开发了一种高AM含量的锰基无序岩盐(Mn-DRXs)正极(>95 wt%-AM),涵盖从固有的材料特性到电极的微观结构,以解决Mn-DRX研究中的艰巨挑战。结果发现,Mn-DRX在高AM浓度电极中的失效源于其极低的电导率(10-10-10-8S/cm)和随着循环过程中体积变化而导致的导电网络破坏。这些失效模式可以通过电渗透工程和电极机械性能的增强来解决,实现几乎全AM的Mn-DRX正极(~96 wt%-AM)和迄今为止报道的最高应用级能量密度(~1050 Wh/kg-正极)。这项工作进一步揭示了Mn含量对Mn-DRX电导率和体积变化的权衡作用,为材料设计提供指导,以推进无Co/Ni的LIB技术。该研究以题目为“Nearly all-active-material cathodes free of nickel and cobalt for Li-ion batteries”的论文发表在国际顶级期刊《Energy & Environmental Science》上。

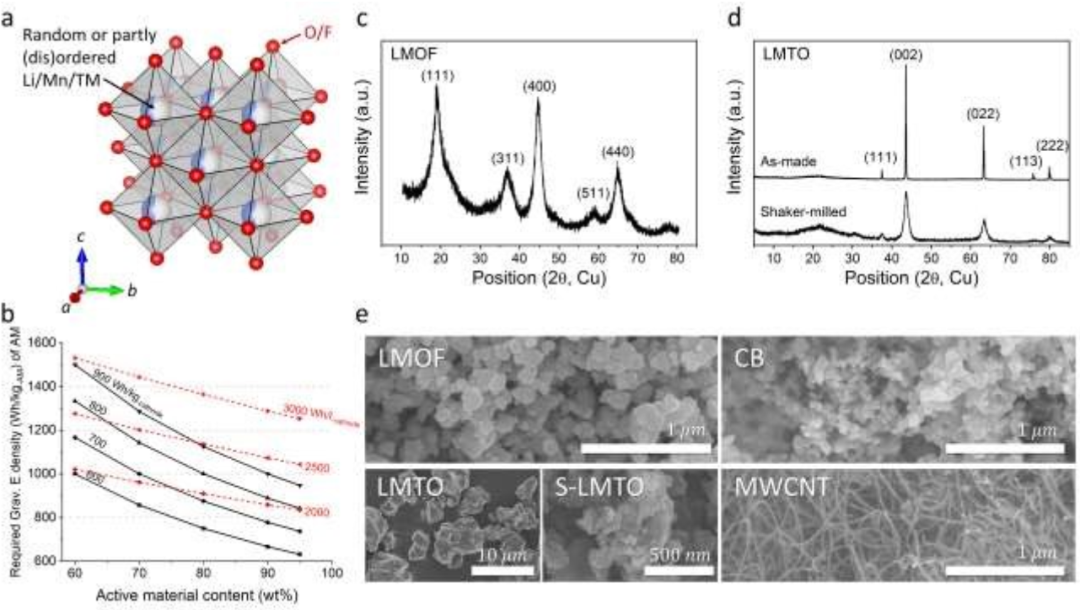
【图1】Mn-DRX正极组分的结构。(a)Mn-DRX的晶体结构示意图。(b)Mn-DRX中实现各种目标正极能量密度(Wh/kg-正极,Wh/l-正极)所需的重力能量密度(Wh/kg-AM)与AM含量的函数关系。(c)LMOF和(d)LMTO(振动球磨前后)的XRD谱图。(e)LMOF、LMTO、振动球磨LMTO(S-LMTO)、炭黑(CB)和多壁碳纳米管(MWCNT)的SEM图像。
本工作选择Li1.68Mn1.60O3.7F0.3(机械化学法制备的LMOF)和Li1.2Mn0.4Ti0.4O2(固态合成和粉碎法制备的LMTO)进行初步试验。图1c和1d的X射线衍射(XRD)图显示,LMOF为部分有序尖晶石(具有类尖晶石局部阳离子有序的DRX),而LMTO为完全阳离子无序的DRX。对制备的LMTO颗粒进行振动球磨,使XRD峰变宽,表明颗粒粉碎,与扫描电镜观察结果一致(图1e)。振动球磨后的LMOF和LMTO均显示出粒径较小的多晶颗粒(直径d<300 nm),这与之前Mn-DRX的研究结果一致。图1e还显示了用于制作正极膜的炭黑碳纳米管和MWCNT的SEM图像。炭黑颗粒具有d<50 nm的球形结构,而MWCNT具有典型的一维纳米管形态。
Mn-DRX在含有过量导电添加剂和粘合剂的稀释电极中循环使用。为了研究它们在AM-浓电极中的行为,分别以CB和PVDF为导电添加剂和粘结剂,以70:20:10 (AM:CB:PVDF, wt%)、80:10:10和90:5:5的比例制备了不同电极组成的LMOF和LMTO薄膜。增加电极中的AM含量会降低电极的厚度,从而获得与LMOF(或任何Mn-DRX)密度(~3.96 kg/l)相似的载量(即mg-正极/cm2)电极。
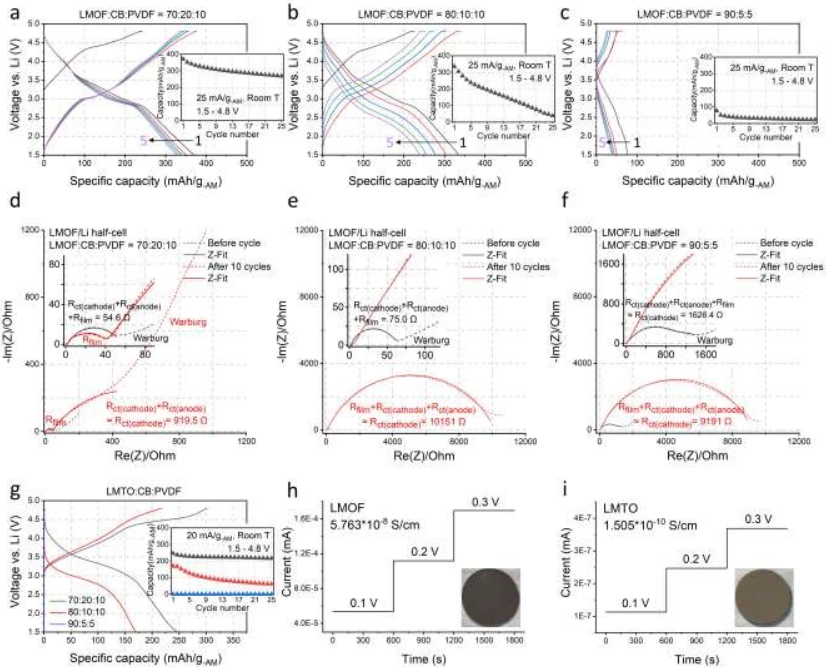
【图2】研究了LMOF和LMTO在不同电极组成下的电化学性能及其电导率。(a-c)在(a)70:20:10(AM:CB:PVDF,按重量计)、(b)80:10:10和(c)90:5:5电极膜中,在25 mA/g-AM和1.5-4.8 V之间循环时LMOF的电压分布和容量保持。(d-f)循环前(黑色)和10次循环后(红色)在1.5-4.8 V之间25 mA/g-AM下(d)70:20:10,(e)80:10:10和(f)90:5:5薄膜(短横线:实验值,线:拟合值):正极/电解质和固体/电解质界面电阻(Rfilm),正极(Rct(正极))或负极(Rct(负极))的电荷转移电阻和Warburg电阻。(g)LMTO在20 mA/g-AM、1.5-4.8 V、70:20:10、80:10:10和90:5:5薄膜下的第一次循环电压分布和容量保持情况。用于电导率测量的(h)LMOF和(i)LMTO的直流极化测试结果:插图显示了该测试中使用的LMOF和LMTO颗粒的图像。
图2a、2b和2c显示了LMOF在70:20:10、80:10:10和90:5:5薄膜(25 mA/g-AM, 1.5-4.8 V,室温)下的电压分布。这些电极的载量始终保持在~5 mg-正极/cm2。在70:20:10的薄膜中,LMOF在第一次放电时提供370 mAh/g-AM,在5次和25次循环后其容量分别下降到327和268 mAh/g-AM。同时,LMOF 80:10:10电极在第1、5和25次循环中提供335、239和38 mAh/g-AM,显示出更小的容量和更快的容量衰减。此外,90:5:5薄膜中的LMOF在第1、5和25个循环中仅达到77、39和23 mAh/g-AM。最后,在没有电极稀释的情况下,在250 mA/g-AM的较高倍率下,LMOF的容量也显著下降,在70:20:10的薄膜中为325 mAh/g-AM,而在90:5:5的薄膜中完全没有容量。
LMOF在AM浓电极中的性能较差是由于电荷转移电阻显著增加。图2d、2e和2f显示了LMOF:CB:PVDF电极在1.5-4.8 V, 25 mA/g-AM下循环前和10次循环后的Nyquist图。对于70:20:10的电极,在高频和中频范围内(凹陷的)半圆,对应于正极和负极的电荷转移电阻(Rct(正极)和Rct(负极))和电极/电解质膜电阻(Rfilm来自正极/电解质界面,CEI和固体-电解质界面,SEI)的直径在循环前为~55 Ω,在循环10次后略有增加到~920 Ω,主要是由于Rct(正极)的变化。在循环之前,使用80:10:10 LMOF薄膜的LMOF/Li电池在相同频率范围内也显示出~75 Ω的小直径半圆,在10次循环后直径增加到接近~10000 Ω(70:20:10的情况下为~920 Ω),这主要是由于LMOF的电荷转移衰减更大,阐明了80:10:10薄膜中LMOF容量更小和容量损失加速的原因(图2b)。对于90:5:5的薄膜,电阻已经在开始时高得多~1600 Ω,在10个循环后达到~9200 Ω(图2f)。LMTO表现出与LMOF相似的行为。当电极组成从70:20:10变化到80:10:10和90:5:5时,LMTO的容量从~250 mAh/g-AM下降到~170和0 mAh/g-AM,表明在浓缩膜中的性能下降比LMOF更严重(图2g)。此外,随着AM含量的增加,LMTO的容量保留率急剧下降。
使用直流极化方法,评估了LMOF和LMTO颗粒的电导率,因为低AM电导率可以限制低CB含量的浓膜中的电荷转移反应(图2h和2i)。LMOF和LMTO的电导率分别为~5.76×10-8S/cm和~1.51×10-10S/cm,明显小于传统的层状氧化物如LiNi0.8Mn0.1Co0.1O2(~1.1 × 10-3S/cm),LMTO的值甚至低于LiFePO4(~5 × 10-8S/cm),特别是,LMTO的电导率比LMOF低~380倍,这与LMTO的颜色比LMOF浅一致(图2h和图2i),这解释了为什么LMTO的性能更多地取决于CB含量而不是LMOF。

【图3】LMOF/CB/PVDF电极循环前后的微观结构。(a-c)循环前(a)70:20:10、(b)80:10:10和(c)90:5:5电极中LMOF(红色)、CB/PVDF(蓝色)和孔(白色)的SEM-EDS图。(d-f)LMOF 80:10:10电极(d)循环前、(e)5次循环后、(f)20次循环后[1.5-4.8 V, 25 mA/g-AM]的SEM图像:LMOF(白色)、CB/PVDF(灰色)、孔隙(黑色)。
循环导电性差的AM需要导电添加剂如CB在电极内的渗透,与AM颗粒形成密切接触。扫描电镜能谱(SEM-EDS)分析证实,炭黑的渗透在AM浓缩膜中受到限制。图3a、3b和3c显示了原始70:20:10、80:10:10和90:5:5电极截面上LMOF(红色)、CB/PVDF(蓝色)和孔隙(白色)的分布。与70:20:10或80:10:10电极不同,90:5:5电极中的CB渗透(蓝色)受到LMOF颗粒(红色)的干扰,这解释了90:5:5电极中明显更高的电荷转移电阻。
虽然AM的低电导率和有限的炭黑渗透解释了AM浓缩电极中LMOF和LMTO的容量较小,但它们在浓缩电极中更快的容量损失主要源于电极在循环过程中的解体(即电化学疲劳),AM浓缩膜中AM/CB和CB/CB接触的降解更为显著(从而破坏电极中的电渗透),并允许形成CEI层。图3d、3e和3f分别是80:10:10 LMOF电极循环前、循环5次和20次(1.5-4.8 V, 25 mA/g-AM)后的截面SEM图像。白色、灰色和黑色区域分别代表LMOF、CB/PVDF和孔隙。在循环之前,电极中的LMOF和CB/PVDF之间可以看到紧密的接触(图3d),但是在5次循环之后,出现了许多孔隙,使LMOF/CB和CB/CB接触松动(图3e)。经过20次循环后,电极的某些区域出现了明显的孔隙堆积(图3f)。此外,观察到循环后裂纹的产生和扩展(图3d-f,底部图像)。
在层状氧化物正极中,由于AM在循环过程中2-3%的体积变化引起电极内的应力和应变,电极通常会因电化学疲劳而解体。然而,某些Mn-DRX的体积变化可达到近10%。特别是,根据XRD分析,LMOF在循环过程中表现出近~8.4%的体积变化,与Ceder等人的数据(~8.3%)一致:请注意,基于XRD的Mn DRX的体积变化分析可能不是最准确的,因为在循环过程中,Mn和O混合氧化还原过程中,来自Mn DRX粉碎颗粒形态和结构非晶化的宽XRD峰,在XRD精修中留下了一定程度的任意性。当电极被稀释时,AM体积变化对AM性能和电极完整性的影响减弱。过量的炭黑和粘结剂可以缓冲应力,在组件之间提供强大的附着力,即使失去一些AM/CB或CB/CB接触也能保持电渗透,并在循环过程中减缓CEI电阻的增长,但这些好处在AM浓缩的电极中消失了。这解释了(i)80:10:10 LMOF电极在循环后出现大裂纹(图3f),而70:20:10电极则没有,以及(ii)CB/PVDF含量较小的非Warburg相关电阻(例如电荷转移)造成的容量损失更快(图2)。
为了克服这些问题,设计了高AM浓度的Mn-DRX正极,用MWCNT代替CB,并使用更多的粘合剂。与CB具有球形纳米颗粒形状(d~50 nm)不同,MWCNT是一维碳(图1e),其电导率(103-105S/cm)高于CB(10-2-10 S/cm)。纳米管结构允许电子沿着管方向(长度>1 μ m)流动而不会中断,与CB相反,CB需要通过CB/CB触点进行远程电子转移,这些触点容易松动或分离(图3f)。此外,每克MWCNT的体积(~4.54 cm3/g)比CB(0.48-0.56 cm3/g)更大,从而允许更少的MWCNT跨越电极进行电渗透。最后,“藤蔓状”MWCNT可以缠绕AM和粘合剂颗粒,以限制电极的分解,从而在AM浓缩的电极中产生更强的电渗透在这方面,MWCNT也被用作其他电极材料的导电添加剂或涂层材料。

【图4】Mn-DRX/MWCNT/PVDF电极的微观结构与性能。(a-b)LMOF(红色)、MWCNT/PVDF(蓝色)和孔(白色)电极的SEM-EDX图谱(左)和LMOF(浅灰色)、MWCNT/PVDF(深灰色)和孔(黑色)在90:5:5 (LMOF:MWCNT:PVDF)电极(a)和(b)循环5次(1.5-4.8 V, 25 mA/g-AM)前后的SEM图像(右)。(c-e)在MWCNT[1.5~4.8 V, 25 mA/g-AM,负极:锂金属]的70:20:10、90:5:5、92:4:4、94:3:3和96:2:2电极中(c)LMTO和(e)LLF在MWCNT[1.5~4.8 V, 20 mA/g-AM,负极:锂金属]的70:20:10、90:5:5、94(LLF):3:3和96:2:2电极中的第一次循环电压分布和容量保持。(f)在含MWCNT的92:4:4高浓度电极中,在(上)25 mA/g-AM[N/P比=1.2,1.0-4.55 V]和(下)0.75 A/g-AM(~4 C)[N/P比=1.2,1.0-4.75 V]条件下,Ti掺杂LMOF(T30和T45)正极下,全电池(负极:石墨)的第一次循环电压分布和容量保持情况。(g)各种Mn-DRX的传统(CB基)电极能量密度(Wh/kg-正极)和MWCNT基电极能量密度与AM含量(%)的函数关系。
图4a和4b显示了用LMOF、MWCNT和PVDF制作的90:5:5电极在1.5-4.8 V, 25 mA/g-AM5次循环前后的EDS图和SEM图像。EDS图中的蓝色区域来自MWCNT和PVDF。与CB的情况不同(图3c),MWCNT/PVDF在整个90:5:5电极中渗透,这有利于电子传递并提高电极的机械强度。反过来,具有MWCNT的LMOF电极显示出非常高的容量和能量密度,>360 mAh/gAM和~1090 Wh/kg-AM(~4330 Wh/l-AM:根据LMOF的理论晶体密度计算,3.96 kg/l),即使AM含量增加到96 wt%(图4c),在30次循环(1.5-4.8 V, 25 mA/g-AM)中,容量保留率(~65%)没有任何明显差异,明显优于80:10(CB):10 LMOF电极,在25次循环后显示<50 mAh/g-AM(~11%保留率)(图2b)。与碳纳米管相比,MWCNT的容量保持能力得到了提高。此外,对于MWCNT,即使是96 wt%-AM的LMOF电极,在2 A/g-AM和4 A/g-AM下,其倍率性能也十分优异,分别提供244 mAh/g-AM和194 mAh/g-AM。值得注意的是,上文所讨论的LMOF(90):MWCNT(5):PVDF(5)和LMOF(96):MWCNT(2):PVDF(2)电极的电极孔隙率分别为13.53%和16.65%,其载量为~5 mg-cathode/cm2。
在循环过程中,仍然观察到90:5(MWCNT):5 LMOF电极(1.5-4.8 V, 25 mA/g-AM,经过5次循环)产生了一些孔,在EDS图中可见白色区域,在SEM图像中可见黑色区域(图4a和4b)。随着LMOF的AM水平问题,这种剩余孔隙度的增加可能构成剩余容量的衰减。同时,与80:10(CB):10 LMOF电极在20次循环后的高度脆性不同,92:4(MWCNT):4电极没有表现出这种脆性,表明即使使用较少的粘合剂也能改善机械性能。
还将MWCNT应用于LMTO和Li1.25Mn0.75O1.33F0.67(LLF)。即使96:2(MWCNT):2 LMTO电极也能提供~220 mAh/g-AM(~673 Wh/kg-AM,~2,560 Wh/l-AM),其容量保持与70:20(MWCNT):10和90:5(MWCNT):5电极(1.5-4.8 V, 20 mA/g-AM)相似,与90:5(CB):5电极显示零容量相反(图4d和2g)。同时,AM含量越高(MWCNT越少),LMTO的第一次放电容量降低比LMOF更为明显,考虑到LMOF在70:20(MWCNT):10和96:2(MWCNT):2电极下的容量降低约7%,而LMTO的容量降低约14%。这一趋势与LMTO的导电性比LMOF差380倍有关,这使得它的容量对碳量更敏感(这里是MWCNT)。
同样,即使在96:2(MWCNT):2电极(图4e)中,LLF也能提供~250 mAh/g-AM(~786 Wh/kg-AM,~3,000 Wh/l-AM)。然而,与LMOF和LMTO不同的是,LLF的容量保留明显变差,分别为94 wt%-AM和96 wt%-AM(图4e)。96 wt%-AM和70 wt%-AM的LLF电极在30次循环后的相对容量保持率仅为~81%,而LMOF和LMTO电极的相对容量保持率分别为~100%和~98%。这似乎是矛盾的,因为LLF(~1.52×10-9S/cm)的电导率比LMTO高10倍,但LLF在循环过程中表现出比LMTO(~4.3%)大得多的体积变化(~7%),从而更明显地促进了电极的分解。有趣的是,LMOF的体积变化(~8.4%)略大于LLF,但MWCNT基LMOF电极在96% wt%-AM和70% wt%-AM的情况下表现出几乎相同的容量保留。这种差异很可能是由于LMOF的导电性比LLF(约38倍)和LMTO(约380倍)高得多,这使得LMOF能够更好地传导电流(除了MWCNT),即使孔隙度增加,也能保持电极的导电性。因此,为了在AM浓缩的电极中保持稳定的容量,Mn-DRX必须具有更高的导电性,较小的体积变化,或者组装在高度稳定的电极复合基质中。
使用92:4(MWCNT):4(PVDF)电极,评估了Mn-DRX正极在石墨负极的全电池中的性能(图4f)。本实验采用掺钛的部分有序LMOF材料Li1.68Mn1.30Ti0.30O3.7F0.3(T30)和Li1.68Mn1.15Ti0.45O3.7F0.3(T45),因为它们比LMOF具有更好的容量/电压保持能力,减少了过渡金属(TM)的溶解,具有更高的库仑效率,因此更适合于全电池测试。在25 mA/g-AM下,在1.0-4.55V(图4f)的全电池中循环时,T30和T45可以达到689 Wh/kg-正极(770 Wh/kg-正极)和675 Wh/kg-正极(737 Wh/kg-正极),这与LiNi0.8Co0.1Mn0.1O2正极在全电池中的能量密度相当(699-732 Wh/kg-正极,取决于上截止电压)。这里报道的T30和T45的比容量和能量密度是基于T30和T45的重量,在全电池中实现高容量所需的化合物预锂化后,其中(与半电池不同)部分有序(最初含有锂空位)化合物在第一次放电期间的过锂化是不可能的,因为使用了不含锂的石墨负极。此外,92:4(MWCNT):4 T30和T45电极在全电池的放电容量分别为202 mAh/g-AM和162 mAh/g-AM,在0.75 A/g-AM(~4C, 1.0-4.75V,图4f,底部)的大电流下,在200次循环中,容量保持率分别为53%和70%。这相当于T30的能量密度为503 Wh/kg-正极(功率密度为670 W/kg-正极),T45的能量密度为386 Wh/kg-正极(功率密度为673 W/kg-正极)。MWCNT的电渗透工程可以恢复Mn-DRX在几乎全AM正极中的容量,实现迄今为止报道的Mn-DRX正极最高能量密度~1000 Wh/kg(图4)。然而,Mn-DRX的体积变化仍然会导致剩余容量损失,如果AM具有更高的导电性(例如,LMOF与LLF),其有害影响较小。此外,AM电导率决定了AM浓缩电极的倍率性能。因此,在限制Mn-DRX体积变化的同时提高导电性是开发实用的全AM Mn-DRX正极的关键。
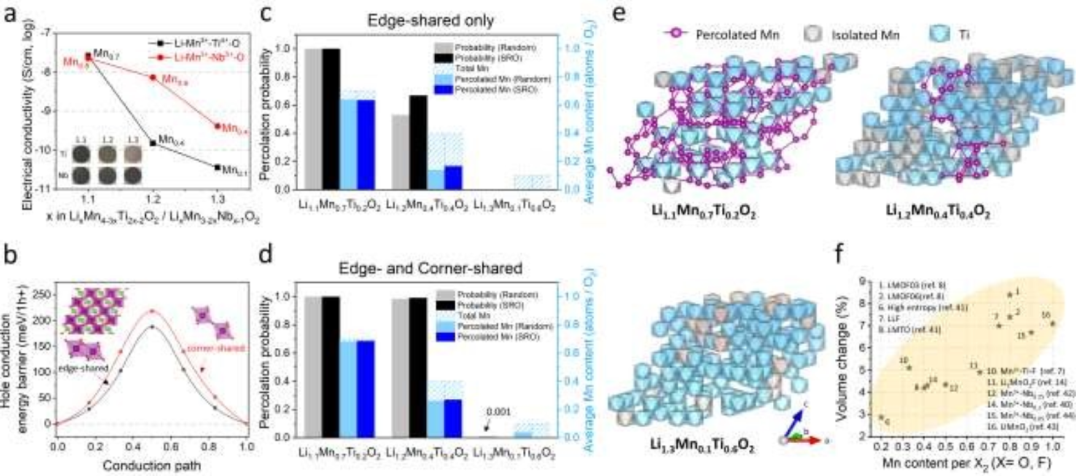
【图5】Mn-DRXs的电导率和体积变化。(a)Li+-Mn3+-Ti4+-O2-和Li+-Mn3+-Nb5+-O2-的电导率(S/cm, log)随锂过量水平(Mn含量水平)的变化。(b)极化子通过共边或共角Mn-Mn路径跳跃时的能量分布。(c,d)通过随机蒙特卡罗(MC)和考虑SRO的马尔可夫链MC(MCMC)模拟得到的(c)仅边共享路径和(d)边和角共享路径,DRX Li+-Mn3+-Ti4+-O2-化合物中Mn的平均渗透概率(~电子渗透概率)和每化学计量量O2的平均渗透Mn含量。(e)Li1.1Mn0.7Ti0.2O2、Li1.2Mn0.4Ti0.4O2和Li1.3Mn0.1Ti0.6O2具有代表性的MC结构。通过边和角共享路径包含在电子渗透网络中的Mn离子用紫色球标记,连接的Mn位点用紫色键桥接。(f)报告的各种Mn-DRX正极的第一次充电体积变化(%,绝对值(体积(第一次充电)-体积(原始))/体积(原始)*100%)作为每X2(X=O, F)Mn含量的函数,除了部分有序的Mn-DRX(LMOF03, LMOF06),其第二次充电体积变化被认为是由于第一次放电时的过锂化。
进一步研究发现,Mn含量与Mn-DRX的电导率有很好的相关性;因此,保持高含量将是有益的。图5a显示了Li+-Mn3+-Ti4+-O2-和Li+-Mn3+-Nb5+-O2-的电导率:注意,这里的趋势比绝对测量值更重要,因为它们可以随着不同的测量技术和颗粒形态而略有变化。当锂过量浓度从10%增加到20%和30%时,Li+-Mn3+-Ti4+-O2-的电导率从2.62×10-8S/cm下降到1.51×10-10S/cm和3.60×10-11S/cm;Li+-Mn3+-Nb5+-O2-的电导率分别为2.29×10-8S/cm~7.35×10-9S/cm和4.12×10-10S/cm。有趣的是,当锂过量时,Li+-Mn3+-Nb5+-O2-表现出比Li+-Mn3+-Ti4+-O2-更高的电导率,当锂过量20%时,电导率提高了约50倍。Ti4+和Nb5+是没有d电子的d0TM物质,这使得它们在正极充电时不能氧化,参与充电诱导的空穴极化子传导。此外,它们几乎不构成Mn3+-DRX的价带或导带,也限制了它们参与热激发电荷载流子传导。因此,电导率的这种差异很可能是由于在相同的锂过量水平下,Li+-Mn3+-Nb5+-O2-中的锰含量高于Li+-Mn3+-Ti4+-O2-。例如,在锂过量20%时,Li+-Mn3+-Nb5+-O2-的Mn含量为Mn0.6(Li1.2Mn0.6Nb0.2O2);而Li+-Mn3+-Ti4+-O2-则为Mn0.4(Li1.2Mn0.4Ti0.4O2)。
这一趋势与我们利用DFT计算和蒙特卡罗(MC)渗流模拟对Mn-DRX导电的理论研究是一致的。导电可以源于离子和电子(电子、空穴)的传递。这里我们考虑充电诱导空穴传输作为主要的导电性来源,因为在正极充电时自然引入空穴。离子传导应该是样品的次要来源。否则,由于Li+通过0-TM渗透网络的扩散得到改善,电导率反而会随着Li过量的增加而增加。
由于Mn-DRX在放电状态下的费米能级位于Mn 3d带内,我们通过连接的Mn位计算了空穴极化子跳跃势垒(图5b)。为了进行计算,制备了一个含有对称边共享和角共享Mn-Mn八面体的阳离子混合LiMnO2模型结构。结果发现,当Mn-Mn八面体是边共享的(~180 meV/h+)而不是角共享的(~220 meV/h+)时,空穴极化子跳势垒较低,这意味着如果晶体结构中有更多的边共享的Mn八面体,Mn-DRXs中的空穴传导会更快。由于沿边共享和边角共享Mn-Mn路径之间的能隙(40 meV/h+)不高,边角共享路径也可能有助于导通。
由于Mn-DRX中的空穴极化子传导得益于连续连接的Mn位,我们采用随机阳离子排序的MC渗流模拟和考虑短程有序(SRO)的马尔可夫链MC(MCMC)渗流模拟研究了两种类型DRX模型结构中Mn位的渗流特性。图5c显示了Li+-Mn3+-Ti4+-O2-中“共享边”Mn位点的渗透概率以及渗透网络中每化学计量O2的平均渗透Mn含量。对于阳离子排序随机的结构,随着Li过量浓度从10%(Li1.1Mn0.7Ti0.2O2)增加到20% (Li1.2Mn0.4Ti0.4O2)和30%(Li1.3Mn0.1Ti0.6O2),渗透概率从100%急剧下降到50%。此外,随着锂过量的增加(Mn含量的降低),渗透网络中每O2的平均Mn含量也显著降低。在模拟中加入“共角”Mn八面体(图5d),Li1.1Mn0.7Ti0.2O2和Li1.2Mn0.4Ti0.4O2的渗流概率都接近100%。然而,Mn在渗透网络中的比例从98%下降到63%,这表明即使考虑了共享边和共享角的Mn-Mn跳跃,充电时Mn上产生的许多空穴也不能直接参与Li1.2Mn0.4Ti0.4O2中的导电,如图5e所示的代表性MC结构所示。同样,考虑SRO的结构也呈现出相同的趋势(图5c和d)。Li+-Mn3+-Ti4+-O2-中凝聚Mn离子略微提高了Mn渗透概率和网络内可达Mn含量,显示出SRO的矛盾效应,该效应被证明对Mn-DRX中Li扩散的0-TM渗透带来了“负面”影响。总的来说,这些结果支持了实验结果,Mn含量与Mn-DRX的电导率有很好的相关性。
虽然增加Mn含量可以提高电导率,但较高的Mn含量通常会增加Mn-DRX在循环过程中的体积变化(图5f),这可能是由于较高的Mn3+含量导致更明显的Jahn-Teller(JT)畸变。例如,Li1.2Mn0.4Ti0.4O2(Mn0.4)、Li1.25Mn0.5Nb0.25O2(Mn0.5)、和Li1.30Mn0.4Nb0.3O2(Mn0.4)的体积变化分别为~4.3%(280 mAh/g-AM充电后)、~4.3%(300 mAh/g-AM充电后)和~4.2%(270 mAh/g-AM充电后),而LiMnO2(Mn1.0)和Li1.05Mn0.9Nb0.05O2(Mn0.9)的体积变化较大,分别为~7.1%(200 mAh/g-AM充电后)和~6.7%(180 mAh/g-AM充电后)。因此,Mn含量较高的Mn-DRX会发生较大的体积变化,当循环大量锂时,这种变化会加剧,例如LMOF(Mn0.8/O1.85F0.15,循环360 mAh/g)或LLF(Mn0.75/O1.33F0.67,循环250 mAh/g)的体积变化大于7%。

该研究展示了几乎全AM含量的Mn-DRX正极(>95 wt%-AM),这比之前文献报道的70~80 wt%-AM范围有了重大进步,实现了迄今为止报道的最高应用级能量密度。这一成就是由于对浓AM Mn-DRX正极的失效机制有了全面的了解,其中包括固有的材料特性和电极微观结构。在浓AM的Mn-DRX正极中观察到的失效可归因于Mn-DRX极低的本征电导率(10-10-10-8S/cm)和电极内孔隙生长和裂纹扩展等电化学疲劳现象。这些问题的产生是由于Mn-DRX的体积变化(>8%)和粉碎形态,最终导致电极内电渗透网络的崩溃,从而限制了它们的电化学性能。为了解决这些问题,引入了一种电极工程策略,利用多功能碳和粘合剂构建一个强大的电渗透网络。这种方法可以实现几乎全AM(96 wt%-AM)的Mn-DRX正极,正极能量密度为~1050 Wh/kg正极。此外,研究发现,增加Mn-DRX中的Mn含量可以提高导电率,但会加剧循环过程中的体积变化。这种权衡强调了通过互补的电极水平工程来解决高度AM浓度的Mn-DRX正极复杂的电化学机械失效的重要性,正如本研究所证明的那样,而不是仅仅依赖于Mn-DRX的成分改性。总的来说,这项工作将Mn-DRX研究推进到更高的技术水平,为开发高能量、几乎全AM的无Co/Ni锂离子正极铺平了道路。

参考文献
Jinhyuk Lee, Eunryeol Lee, Dae-Hyung Lee, Stephanie Bessette, Sang-Wook Park, Nicolas Brodusch, Gregory Lazaris, Hojoon Kim, Rahul Malik, Raynald Gauvin* and Dong-Hwa Seo*. Nearly all-active-material cathodes free of nickel and cobalt for Li-ion batteries. Energy & Environmental Science.
DOI:10.1039/D4EE00551A
https://doi.org/10.1039/D4EE00551A
