

1、什么是MDR认证?
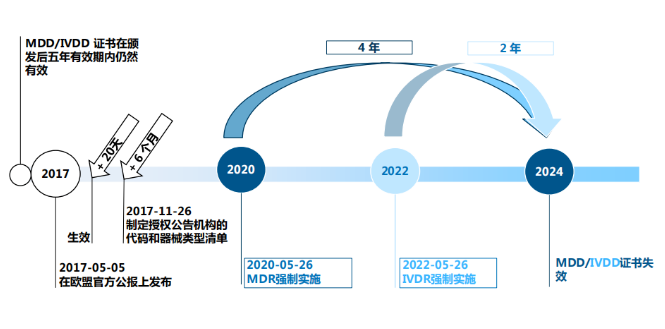
医疗器械法规 MDR(Medical Device Regulation(EU) 2017/745)2017年5月5日,欧盟官方期刊(Official Journal of the European Union)正式发布了欧盟关于医疗器械第2017/745号法规(简称“MDR法规”)。MDR法规于2017年5月26日正式生效,并于2021年5月26日正式取代原有的医疗器械指令 (MDD 93/42/EEC) 和有源植入性医疗器械指令(AIMDD 90/385/EEC)。

体外诊断医疗器械法规 IVDR (In Vitro Diagnostic Medical Devices Regulation 2017/746/EC)体外诊断试剂及器械的全新法规IVDR将于2022年5月26日强制执行,替代原有的体外诊断。IVDR和IVDD相比最大的变化是产品分类的变化。IVDR按照风险等级将IVD产品分为四类:class A, class B, class C和class D(风险由低到高)。IVDR规定B, C, D类的体外诊断试剂都需要公告机构参与认证。
注意:MDR 法规第 1 条(第 7 点)明确了 IVDR 与MDR 法规之间的关系, 即如果设备同时具有 IVD 组件和其他医疗设备组件,则该设备的 IVD 部件均受 IVDR 法规的约束,其余部分受 MDR 的约束。
2、 MDR的过渡期
3、 MDR的主要变化


强化制造商的责任:
合规负责人

技术文件
1 产品的总体描述 General description of the device | 11.风险分析 Risk analvsis |
2 预期用途描述 Description of intended use | 12.材质说明 Specification of materials |
3 产品等级划分,所选分类规则、理由 Class of device,chosen classification rule and justification | 13.产品外观及内部结构图(包括电路图等) Drawings and schematics |
4.附件描述(如适用) Description of accessories (if applicable) | 14.标识 Labeling |
5. 共同作用药物描述(如适用) Description of incorporated pharmaceutical substances (if applicable) | 15. 包装描述 Description of packaging |
6.所用动物组织描述(如适用)Description of utilized tissues of animal origin (if applicable) | 16. 使用说明 Instructions for use |
7.计划制造方法 Planned production methods | 17.寿命和/或保存期限 Lifetime and/or shelf-life |
8.制造过程各阶段及最终的检验测试Description of checks during and at the end of production | 18.灭菌有效性确认(如适用 Sterilization validation (if applicable) |
9.基本要求事项对照 Responses to essential requirements | 19.测试报告(电气部分、机械部分及生物适应性等方面Test reports (electrical / mechanical / biocompatibility etc.) |
10.适用标准清单 List of applied standards | 20.临床数据 Clinical data |
MDR医疗器械技术资料

财务及保险

相关方的监督



4、MDR医疗器械产品分类

医疗器械法规附录九中详定22条规则,按医疗产品的危险程度,将产品分为Ⅰ类、Ⅱa类、Ⅱb类、Ⅲ类。
产品分类规则:
1、规则应用由器械的预期使用目的决定;
2、如果器械是和其它器械配合使用,分类规则分别适用于每种器械;
3、附件可以和其它一起使用的器械分开单独分类;
4、启动或影响某种器械的软件与器械属于同一类型。
分类准则:
时 间:暂时 (<60分钟)、短期(<30天)、长期(>30天)
创伤性:非创伤、通过孔径创伤,外科创伤 、植入。
适用位置:中央循环、中枢神经系统,其它地方。
能量供应:无源,有源。
5、MDR医疗器械认证流程

MDR医疗器械认证流程
